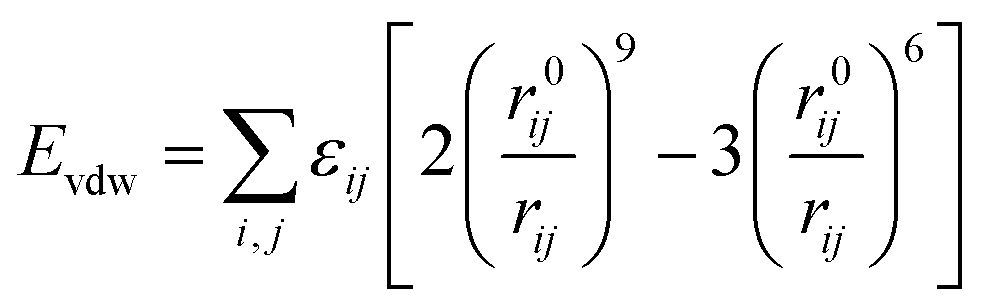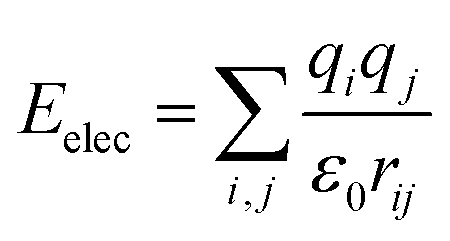DOI:
10.1039/D4RA02964J
(Paper)
RSC Adv., 2024,
14, 21425-21431
Analysis of the adhesion mechanism of functionalized carbon nanotubes by molecular dynamics simulation
Received
22nd April 2024
, Accepted 30th June 2024
First published on 8th July 2024
Abstract
Although a lot of research has been carried out on the adhesion mechanism of gecko bristles, the research on materials inspired by gecko bristles is still limited to the design of geometric structure and the optimization of preparation process, and the adhesion mechanism of materials is still unclear. In this paper, the molecular structure of the end of the bristle-like material is focused on, and the interaction between functional group modified carbon nanotubes and the interface is analyzed by molecular dynamics simulation. Thus, the influence of different polar functional groups on the interfacial force between carbon nanotubes and silica is revealed, and the adhesion enhancement mechanism of polar groups on the interface between carbon nanotubes and silica is further verified.
1. Introduction
Geckos can walk or climb on the surface of walls, ceilings, glass and other materials using the strong adhesion of bristles on their toes. Scientists have put forward many hypotheses to explain the strong adhesion of gecko bristles, including sucker force, electrostatic force, micro-interlocking force, capillary force, van der Waals (vdW) force and so on.1–5 For example, Autumn et al. confirmed that the adhesion of gecko bristles comes from the intermolecular force between keratin molecules of bristles and the contact surface, that is, the van der Waals force mechanism.2,3 In recent years, Alibardi et al. proposed that the adhesion of gecko sole bristles comes from the joint action of van der Waals force and electrostatic force, which is related to the biological tissue and chemical composition of the bristle surface.6–8 With the adhesion characteristics of gecko bristles revealed by many scientists, a series of adhesion materials inspired by the structure of gecko bristles have also been developed, mainly including polymer arrays and vertically aligned carbon nanotubes (VACNT) arrays.9–12 VACNT arrays not only have the advantages of high adhesion strength, no damage to contact surface, and low requirements for use environment, but also have higher elastic modulus, higher thermal conductivity and thermal stability, and good electrical and electrochemical properties, so they have a wide application prospect in electronic devices, wall climbing robots, aerospace and other fields.12–14
Although a lot of research has been carried out on the adhesion mechanism of gecko bristles, the research on adhesion materials is still limited to the design of geometric structures and the optimization of preparation process, and its adhesion mechanism is still unclear, and in particular the influence of terminal molecular composition on its adhesion is rarely studied.15–17 As reported in our recent publications (ref. 18–20), we have used different plasma treatment processes to modify the surface of VACNT arrays and analyzed the influence of surface chemical composition on its adhesion, thus preliminarily revealing that grafting polar functional groups can effectively enhance the adhesion of VACNT arrays through experiments. However, the microscopic mechanism caused by molecular structure is difficult to be obtained by experimental methods, and the specific adhesion enhancement mechanism has not been clearly explained. Therefore, this paper proposes to analyze the interaction between VACNT arrays modified by different functional groups and the interface by molecular dynamics simulation. As described in the reference, the adhesion between VACNT array and interface in the experiment mainly comes from the interfacial adsorption between the sidewall of CNTs on the surface of VACNT arrays and the glass surface. So, the adsorption energy between CNTs with different functional groups and the interface of silica (SiO2) can be calculated by molecular dynamics simulation to analyze the adhesion enhancement mechanism.
Based on the previous experimental research, this paper proposes a molecular dynamics simulation method to study the adhesion mechanism between CNTs and SiO2 interface before and after modification with different functional groups. In this paper, the interaction between CNT modified by fluorine-containing group (–F), carboxyl group (–COOH) and amino group (NH2) and SiO2 interface is discussed respectively, so as to obtain the adhesion mechanism of functionalized VACNT arrays.
2. Experimental
2.1 Molecular dynamics simulations
Molecular mechanics (MM) and MD simulations have been carried out using a commercial software package called materials studio (MS) developed by Accelrys Software, Inc. The interaction between CNTs and SiO2 interface were calculated in the condensed-phase optimized molecular potential for atomistic simulation study (COMPASS) force-field using Forcite module of MS.21,22 The COMPASS potential function calculation includes all bonding energy and non-bonding energy, which is more accurate than general force field, so it is more suitable for this research work.23,24 In the COMPASS force-field, the total potential energy (E) of a molecular system includes the following terms:25| | |
E = Evalence + Enonbond + Ecrossterm
| (1) |
| | |
Evalence = Estretch + Ebend + Etorsion + Einversion + EUB
| (2) |
| | |
Enonbond = Evdw + Eelec + EH
| (3) |
| |
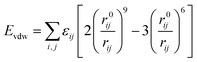 | (4) |
| |
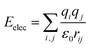 | (5) |
The total potential energy (E) of a molecular system includes a valence energy (Evalence), a nonbond interacting energy (Enonbond), and a crossterm interacting energy (Ecrossterm). The valence energy (Evalence) generally includes a bond stretching term (Estretch), a bond bending term (Ebend), a dihedral bond-torsion term (Etorsion), an inversion (or an out-of-plane interaction) term (Einversion), and an Urey–Bradley (involves interactions between two atoms bonded to a common atom) term (EUB). The nonbond interacting energy (Enonbond) includes a van der Waals interaction energy (Evdw), an electrostatic interacting energy (Eelec), and a hydrogen bonding energy (EH). Atom i is the charge acceptor, atom j is the charge donor, qi, qj are the charges of two atoms. Where rij is the distance between two atoms, r0ij is the atomic distance when the action potential is zero, εij is the lowest value of the action potential, and ε0 is the coefficient.
2.2 Interaction between CNT and SiO2 substrate
Firstly, the SiO2 model is established in MS software, and the model is changed into a rectangular cell to facilitate the subsequent calculation. After expanding the unit cell, the unit cell is sliced along the (001) direction, a vacuum layer with a height of 50 Å is added, and the end cap is hydrogenated. On the one hand, the uncapped silica is negatively charged, and CNT will stand up after calculation, so that the hydrogen end is close to the surface of SiO2. On the other hand, the surface of SiO2 is often partially hydroxylated by environmental influence in experiments. The structure of the SiO2 model is shown in Fig. 1, in which the cell parameters are a = 42.54 nm, b = 49.13 nm, c = 66.72 nm, α = β = γ = 90°, with a total of 3560 atoms.
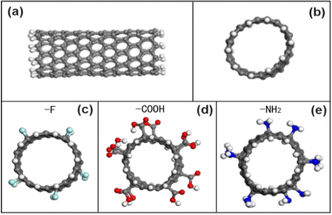 |
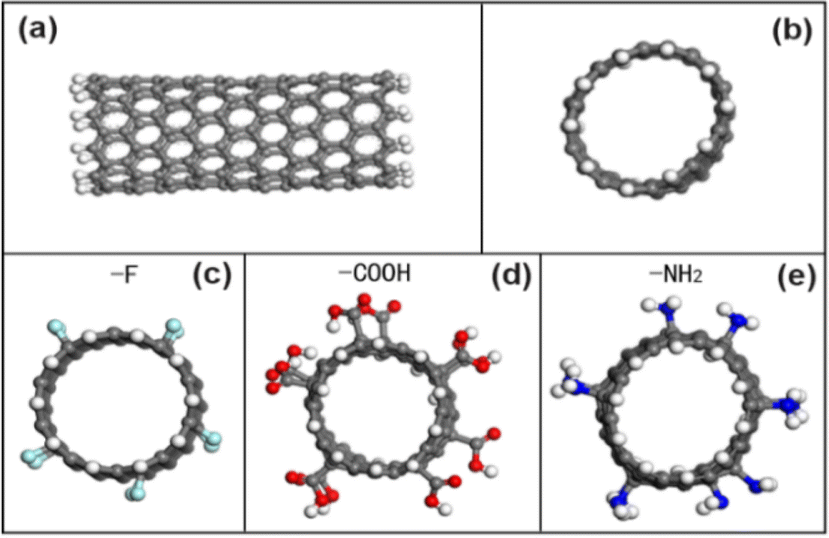
| | Fig. 1 (a) The side view of an unmodified CNT model; (b) the cross-sectional view of an unmodified CNT model; (c) the CNT model of grafted with F atoms; (d) the CNT model grafted with COOH groups; (e) the CNT model of grafted with NH2 groups. | |
Then, the CNT (sawtooth) model is established in the build module of MS software, and the structural index is set to N = 10, M = 0, and 5 carbon ring unit periods, and the periodicity is removed and the end cap is hydrogenated. For CNTs modified with different functional groups, functional groups are manually added according to the atomic number ratio of 5% (ratio of functional groups to carbon atoms) to obtain CNTs modified with fluorine-containing groups (–F), carboxyl groups (COOH) and amino groups (NH2), which are labeled as CNT–F, CNT–COOH and CNT–NH2 respectively. Fig. 1 is a model diagram of the initial state of carbon nanotubes (10, 0) before and after modification with different functional groups.
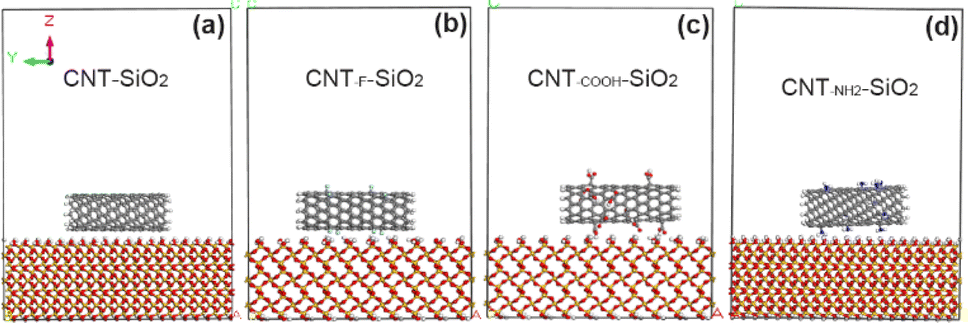
Finally, CNT models with different functional groups were added to the model of SiO2 and moved to the middle position (Fig. 2). All atomic coordinates except the outermost atoms in SiO2 are fixed to reduce the amount of calculation. As reported in the existing literature (Hu26 and Qu27 etc.), the tangled fragments at the end of VACNT array make the CNTs sidewall and the contact surface form a “line” contact, which provides a larger contact area and produces a greater tangential adhesion. Therefore, the adhesion mechanism between the carbon nanotube array and the interface can be revealed by analyzing the interaction between the sidewall and the interface.
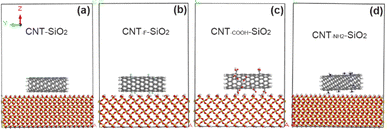 |
| | Fig. 2 Initial model of interface systems between carbon nanotubes with different functional groups and SiO2. (a) Unmodified CNT; (b) the CNT grafted with F atoms; (c) the CNT grafted with COOH groups; (d) the CNT grafted with NH2 groups. | |
The Forcite module of MS software is used to simulate the dynamics of the contact system model, and the canonical ensemble, which can also be called NVT ensemble (that is, the atomic number N, volume V and temperature T of the system remain constant under this ensemble, and the total momentum of the system is zero) is used to control the simulated temperature of the system at 298 K. Before the calculation, the smart algorithm is used to optimize the geometry. The essence of this algorithm is to combine Steepest descent, ABNR and Quasi-Newton methods to eliminate the unreasonable contact between atoms, so as to reduce the amount of calculation and make the results more accurate. Then, the dynamic calculation of the optimized model is carried out, and the time step is set to 2 fs, and the total calculation time is 2000 ps. In the calculation process, the bond length is fixed by LINCE algorithm. In the non-chemical bond action, van der Waals force belongs to short-range force, and its action range is very small, while electrostatic force is a long-range force, and the cutoff distance is always 7.5–12 Å in the calculation of long-range force.28 As reported in the literature, the most computationally tractable treatment of nonbonded interactions in MD utilizes a spherical distance cutoff (typically, 8–12 Å) to reduce the number of pairwise interactions.29 Therefore, the cutoff radius of cutoff distance is set to 10.5 Å in this work. van der Waals interaction adopts atom based algorithm, which is often used to directly calculate the non-bond interaction energy between atoms. Ewald algorithm is used in electrostatic interaction, which is a common method to calculate the long-range electrostatic interaction energy in periodic systems. The calculation formulas of adsorption energy Eadhesion at the interface between different CNT and SiO2 in simulation systems are as follows:
| | |
Eadhesion = ECNT-SiO2 − ECNT − ESiO2
| (6) |
where
Eadhesion is the adsorption energy between CNT and interface,
ECNT-SiO2 is the total energy of the interface system composed of CNT and SiO
2,
ECNT is the single point energy of CNT after removing all atoms of SiO
2, and
ESiO2 is the single point energy of SiO
2 after removing all atoms of CNT. Here, the CNT in the formula represents different carbon nanotubes, including unmodified CNT, CNT
–F, CNT
–COOH and CNT
–NH2. Finally, the average atomic distance between interfaces can be calculated by VESTA software.
30,31
3. Result and discussion
3.1 Interaction between fluorine-modified CNT and SiO2 substrate
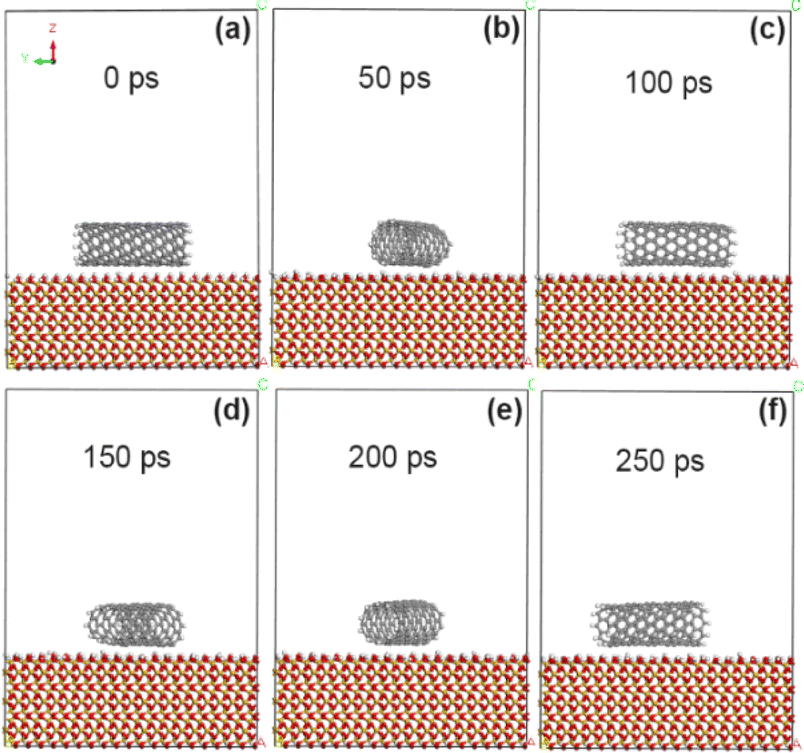
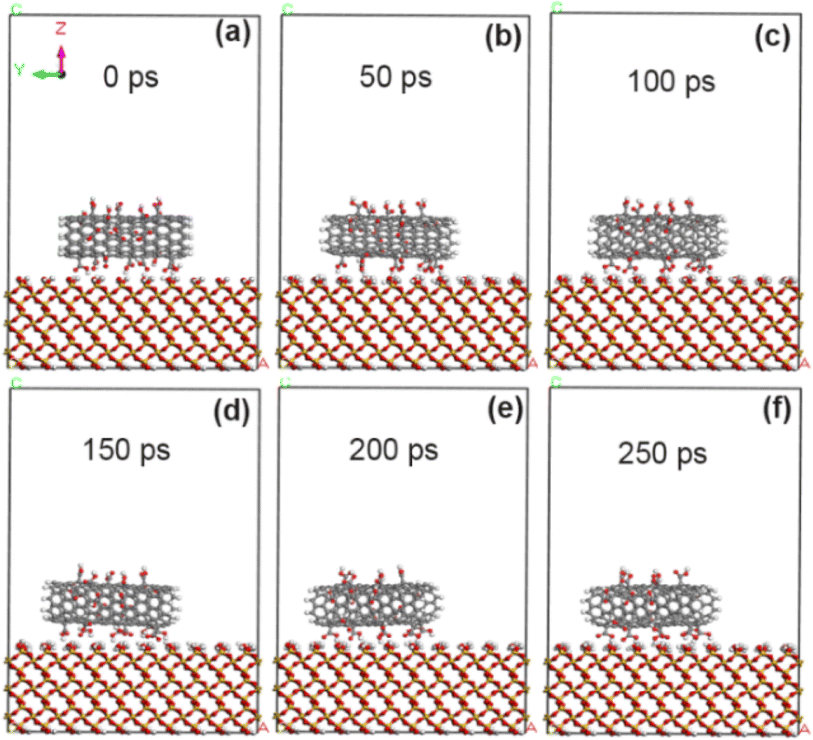
As reported in our previous experiments (ref. 18–20), the grafting amount of fluorine, oxygen and nitrogen atoms by exposure to plasma is in the range of 0–15%. In the simulation calculation, the grafting amount is 5% and 10% in the above range respectively, so that it is convenient to compare the grafting effects of different functional groups. In order to compare the changes of adsorption energy between CNT and SiO2 interface before and after modification with fluorine-containing groups, the adsorption energy between the original CNT and the interface between CNT and SiO2 modified with fluorine-containing groups (–F) was calculated by molecular dynamics simulation. Fig. 3 shows the morphological changes of the original CNT (10, 0) on the SiO2 interface at different times. As the simulation time goes on, the CNT approaches the SiO2 surface and rotates rapidly (at least two times at 250 ps), showing extremely unstable dynamic changes. Combined with the calculation results in Tables 1 and 2, it is concluded that the adsorption energy between unmodified CNT and SiO2 interface is about −53.8 kcal mol−1, and it mainly comes from the non-chemical bonding energy (−55.5 kcal mol−1), in which the van der Waals force energy accounts for the main part (−47.6 kcal mol−1), while the electrostatic force energy is very small, only −1.6 kcal mol−1. The shortest contact distance between carbon atoms on CNT and SiO2 interface (the average distance of four pairs of atoms) is about 3.15 Å by VESTA software.30,31
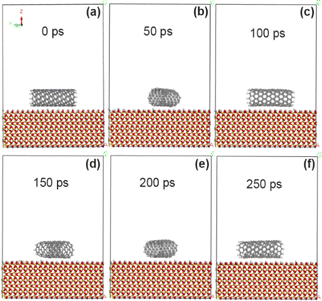 |
| | Fig. 3 State of unmodified CNT on SiO2 surface at different time. (a) 0 ps; (b) 50 ps; (c) 100 ps; (d) 150 ps; (e) 200 ps; (f) 250 ps. | |
Table 1 Interaction of CNT-SiO2 interface before and after fluorine modification (kcal mol−1)
| |
ECNT (–F) |
ESiO2 |
ECNT-SiO2 |
Eadhesion |
| Unmodified |
9313.1 |
−18382.8 |
−9123.5 |
−53.8 |
| –F (5%) |
7264.5 |
−28928.0 |
−21723.7 |
−60.2 |
| –F (10% |
5548.3 |
−17277.5 |
−11792.0 |
−62.8 |
Table 2 Non-chemical bond interaction and interface distance of CNT-SiO2 interface before and after fluorine modification
| |
Enonbond (kcal mol−1) |
Evdw (kcal mol−1) |
Eelec (kcal mol−1) |
Distance (Å) |
| Unmodified |
−55.5 |
−47.6 |
−1.6 |
3.15 |
| –F (5%) |
−63.5 |
−45.7 |
−10.9 |
3.11 |
| –F (10%) |
−65.7 |
−45.1 |
−9.5 |
3.18 |
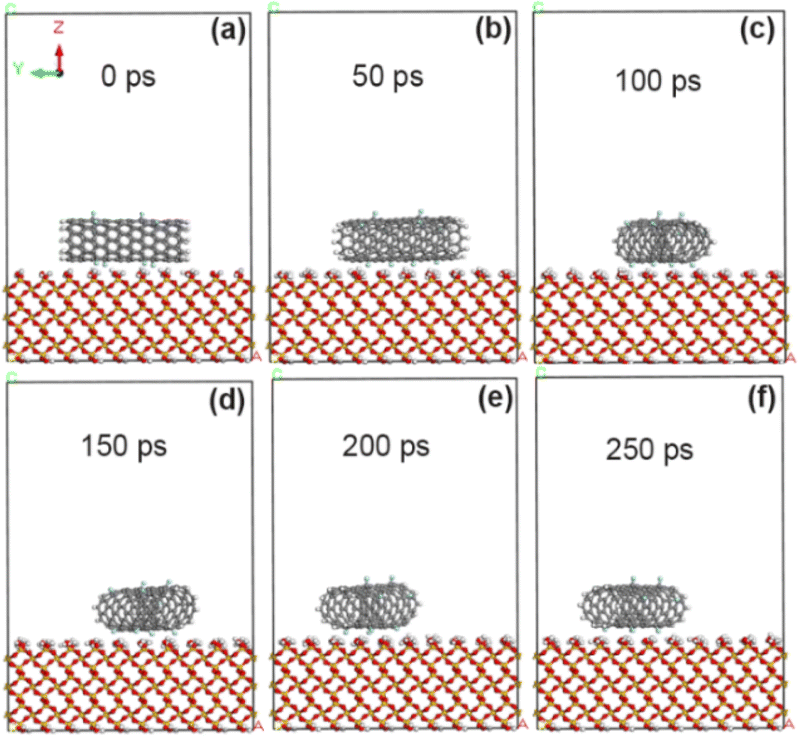
In order to compare the effects before and after functional group modification, fluorine atom (–F) was added to the CNT model according to the grafting amount of 5% of F/C atom percentage, and then geometric optimization and simulation calculation were carried out. As shown in Fig. 4, fluorine-modified CNT at 100 ps produced a small amount of deflection on the surface of SiO2 substrate, and reached a basically stable adsorption state at 150 ps. Combined with the calculation results in Tables 1 and 2, the adsorption energy at the interface between fluorine-modified CNT and SiO2 substrate is improved (from −53.8 kcal mol−1 to −60.2 kcal mol−1). As reported in ref. 18, our previous experimental results show that, the untreated VACNTs exhibit the adhesion strength of 21.1 N cm−2, while the sample after CF4 plasma treatment (XPS data showed that polar fluorine-containing functional groups were introduced) shows the higher adhesion strength of 29.3 N cm−2. Therefore, both theoretical calculation results and experimental results show that the introduction of polar fluorine-containing functional groups can increase the adhesion between carbon tubes and glass interface, and theoretical calculation results reveal that the mechanism is mainly from the enhancement of electrostatic interaction. Among them, the non-chemical bond action energy is about −63.5 kcal mol−1, the van der Waals force action energy has little change, but the electrostatic action energy is obviously enhanced (−10.9 kcal mol−1). At this time, the average distance between carbon atoms on CNT and SiO2 interface atoms is about 3.11 Å, which has little change compared with that before modification.
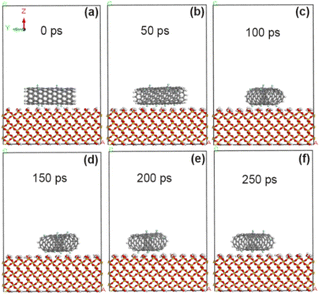 |
| | Fig. 4 State of CNT grafted with 5% fluorine groups on SiO2 surface at different time. (a) 0 ps; (b) 50 ps; (c) 100 ps; (d) 150 ps; (e) 200 ps; (f) 250 ps. | |
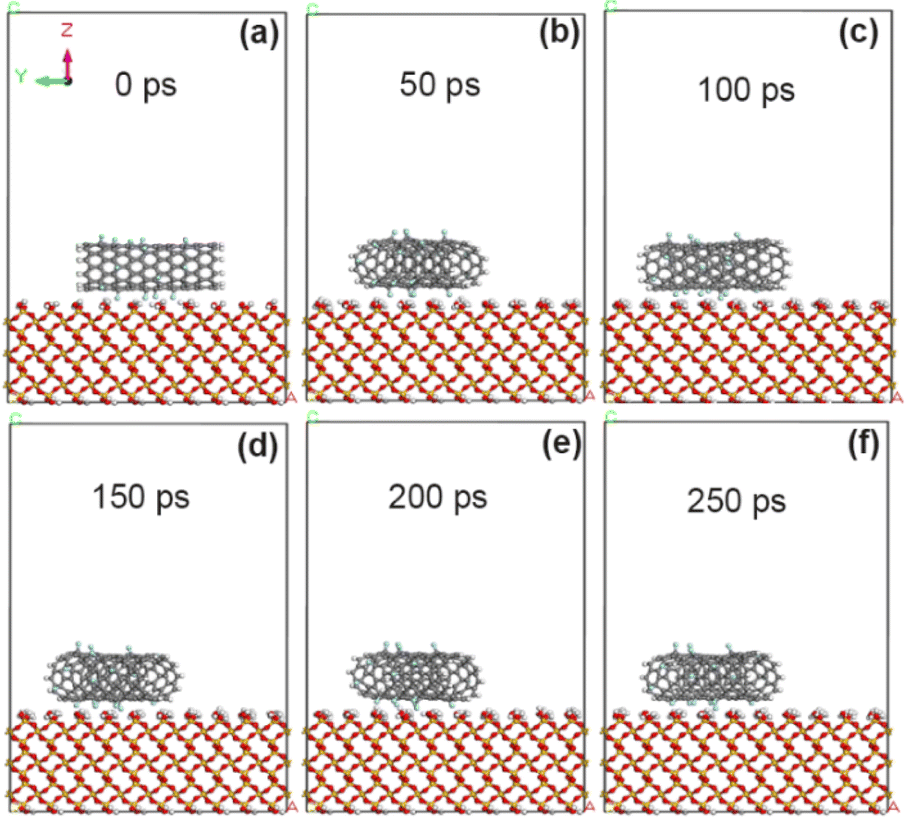
In order to analyze the influence of different grafting amounts of functional groups, fluorine atoms (–F) were added to the original CNT model according to the grafting amount of 10% F/C atom percentage. As shown in Fig. 5, CNT grafted with fluorine atom produces a small amount of deflection on the surface of SiO2 within 50 ps, and has reached a basically stable adsorption state at 100 ps. Combined with Tables 1 and 2 and it is calculated that the adsorption energy of the interface between 10% fluorine-modified CNT and SiO2 is further increased (−62.8 kcal mol−1), and the non-chemical bonding energy is further enhanced (−65.7 kcal mol−1). However, the van der Waals force energy (−45.1 kcal mol−1) and electrostatic energy (−9.5 kcal mol−1) decreased slightly, which may be due to the slight increase of interface distance.
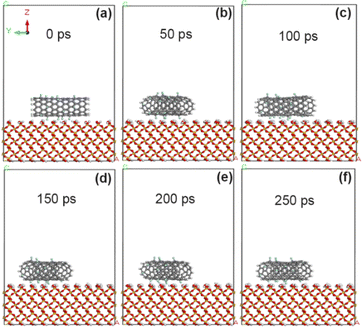 |
| | Fig. 5 State of CNT grafted with 10% fluorine groups on SiO2 surface at different time. (a) 0 ps; (b) 50 ps; (c) 100 ps; (d) 150 ps; (e) 200 ps; (f) 250 ps. | |
3.2 Interaction between carboxyl-modified CNT and SiO2 substrate
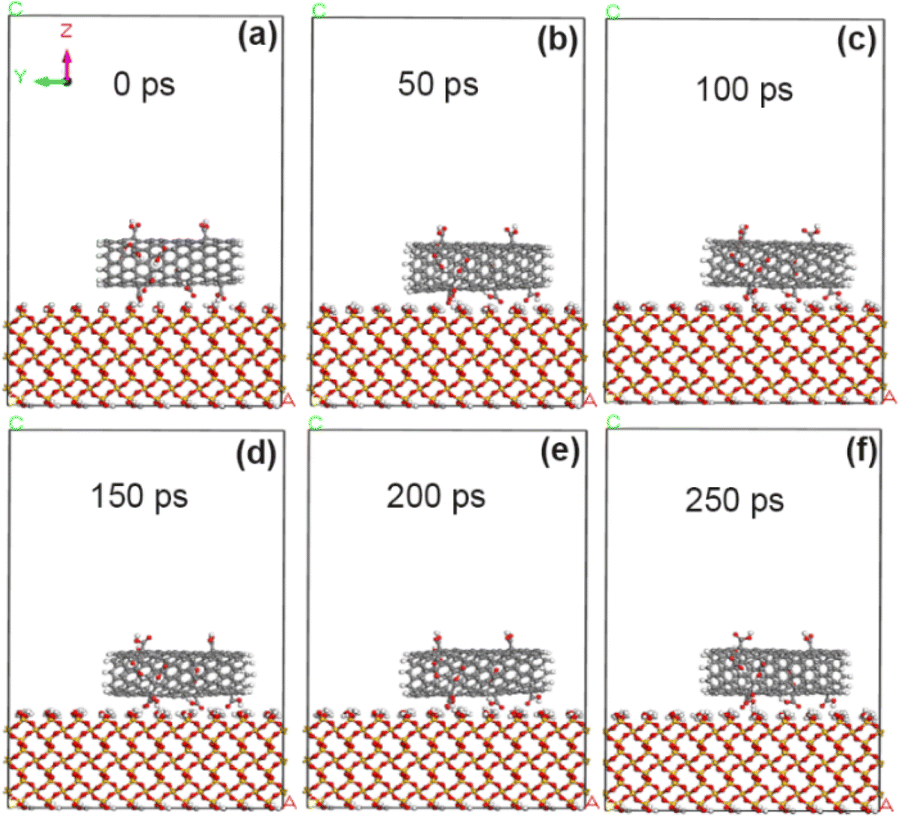
As reported in ref. 19, the experiment reveals that carboxyl groups with strong polarity can significantly improve the adhesion of VACNT arrays. In order to reveal the adhesion enhancement mechanism, here, the carboxyl groups (–COOH) were added to the original CNT model with 5% grafting amount, and then the simulation calculation was made. As shown in Fig. 6, the CNT modified by carboxyl groups within 250 ps did not undergo any rotation movement, and was always stably adsorbed on the surface of SiO2. The calculation results show (Tables 3 and 4) that the adsorption energy of the interface between carboxyl-modified CNT and SiO2 is −69.7 kcal mol−1, which is more obvious than the adhesion enhancement effect of fluorine atom modification (−60.2 kcal mol−1). This is because the carboxyl group with the negatively charged oxygen,32,33 which greatly enhances the electrostatic interaction (the electrostatic interaction energy is enhanced from 1.6 kcal mol−1 to −31.8 kcal mol−1). In addition, the non-chemical bonding energy at the interface between 5% carboxyl-modified CNT and SiO2 increased to −74.6 kcal mol−1, in which the electrostatic interaction energy was significantly enhanced (−31.8 kcal mol−1), while the van der Waals force energy was reduced (−35.3 kcal mol−1). This is because the action distance of van der Waals force is small (suddenly decreased within 3–5 Å), and the carboxyl group with large geometric structure hinders the contact between CNT and interface, which increases the interface distance (3.45 Å) and weakens the action of van der Waals force. However, the action distance of electrostatic force is large (micron level), so this obstacle has relatively little influence on electrostatic action.
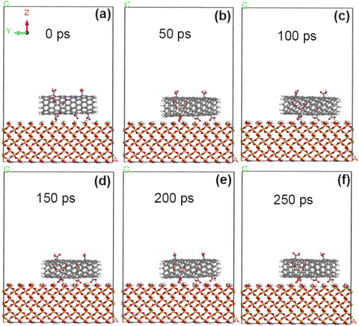 |
| | Fig. 6 State of CNT grafted with 5% carboxyl groups on SiO2 surface at different time. (a) 0 ps; (b) 50 ps; (c) 100 ps; (d) 150 ps; (e) 200 ps; (f) 250 ps. | |
Table 3 Interaction of CNT-SiO2 interface system before and after carboxyl modification (kcal mol−1)
| |
ECNT (–COOH) |
ESiO2 |
ECNT-SiO2 |
Eadhesion |
| Unmodified |
9313.1 |
−18382.8 |
−9123.5 |
−53.8 |
| –COOH (5%) |
3699.8 |
−18374.7 |
−14744.6 |
−69.7 |
| –COOH (10%) |
2200.9 |
−18453.2 |
−16323.7 |
−71.4 |
Table 4 Non-chemical bond interaction and interface distance of CNT-SiO2 interface before and after carboxyl modification
| |
Enonbond (kcal mol−1) |
Evdw (kcal mol−1) |
Eelec (kcal mol−1) |
Distance (Å) |
| Unmodified |
−55.5 |
−47.6 |
−1.6 |
3.15 |
| –COOH (5%) |
−74.6 |
−35.3 |
−31.8 |
3.45 |
| –COOH (10%) |
−77 |
−21.1 |
−42.1 |
3.78 |
After adding 10% carboxyl groups according into CNT, geometric optimization and simulation calculation were carried out again. As shown in Fig. 7, the carboxyl-modified CNT was always stably adsorbed on the surface of SiO2. The calculation results show (Table 3) that the adsorption capacity of the interface between CNT and SiO2 modified by 10% carboxyl group can be further enhanced to −71.4 kcal mol−1, indicating that increasing the grafting amount of carboxyl groups will make the adhesion enhancement effect more obvious. Table 4 shows that the non-chemical bond interaction between 10% carboxyl modified CNT and SiO2 interface can be further improved to −77 kcal mol−1, which is mainly due to the electrostatic interaction energy (−42.1 kcal mol−1). At this time, the interface distance between CNT and SiO2 increased to 3.78 Å, which made the van der Waals force decrease more obviously (−21.1 kcal mol−1).
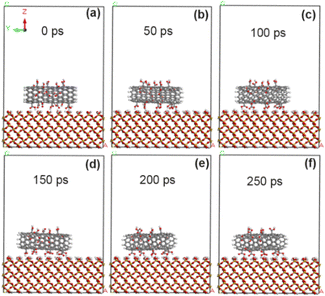 |
| | Fig. 7 State of CNT grafted with 10% carboxyl groups on SiO2 surface at different time. (a) 0 ps; (b) 50 ps; (c) 100 ps; (d) 150 ps; (e) 200 ps; (f) 250 ps. | |
3.3 Interaction between amino-modified CNT and SiO2 substrate
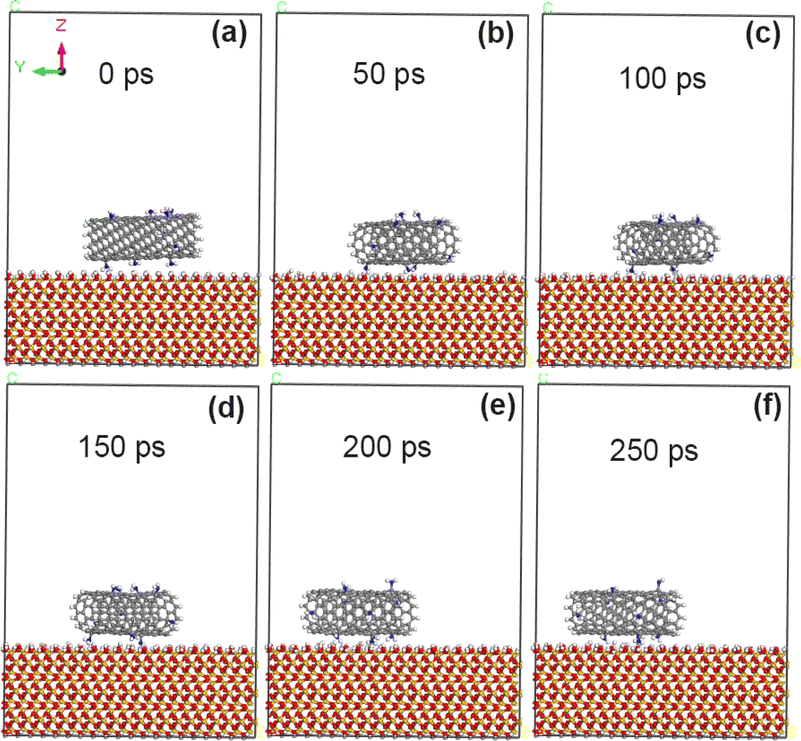
As shown in Fig. 8, when 5% amino groups were grafted on the original CNT model, the adsorption morphology of CNT on the surface of SiO2 changed little, and CNT was always stably adsorbed on the surface of SiO2. Combined with the calculation results in Table 5, it is concluded that the adsorption energy of the interface between amino-modified CNT and SiO2 can reach −82.8 kcal mol−1, indicating that the adhesion enhancement of NH2 is more obvious than that of F and COOH groups under the same grafting amount. The calculated data in Table 6 show that the non-chemical bonding energy between 5% amino modified CNT and SiO2 can reach −87.5 kcal mol−1, of which the electrostatic force can reach −35.5 kcal mol−1 and the van der Waals force can reach −45.1 kcal mol−1.
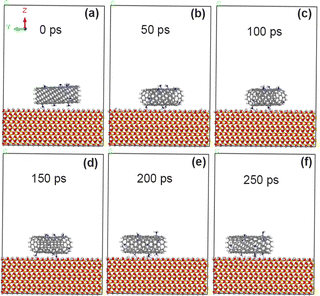 |
| | Fig. 8 State of CNT grafted with 5% amino groups on SiO2 surface at different time. (a) 0 ps; (b) 50 ps; (c) 100 ps; (d) 150 ps; (e) 200 ps; (f) 250 ps. | |
Table 5 Interaction of CNT-SiO2 interface system before and after amino modification (kcal mol−1)
| |
ECNT (–NH2) |
ESiO2 |
ECNT-SiO2 |
Eadhesion |
| Unmodified |
9313.1 |
−18382.8 |
−9123.5 |
−53.8 |
| –NH2 (5%) |
3805.3 |
−18380.9 |
−14658.4 |
−82.8 |
| –NH2 (10%) |
2688.6 |
−7992.1 |
−5381.1 |
−77.6 |
Table 6 Non-chemical bond interaction and interface distance of CNT-SiO2 interface before and after amino modification
| |
Enonbond (kcal mol−1) |
Evdw (kcal mol−1) |
Eelec (kcal mol−1) |
Distance (Å) |
| Unmodified |
−55.5 |
−47.6 |
−1.6 |
3.15 |
| NH2 (5%) |
−87.6 |
−45.1 |
−35.5 |
3.06 |
| –NH2 (10%) |
−81.2 |
−37.9 |
−30.6 |
3.25 |
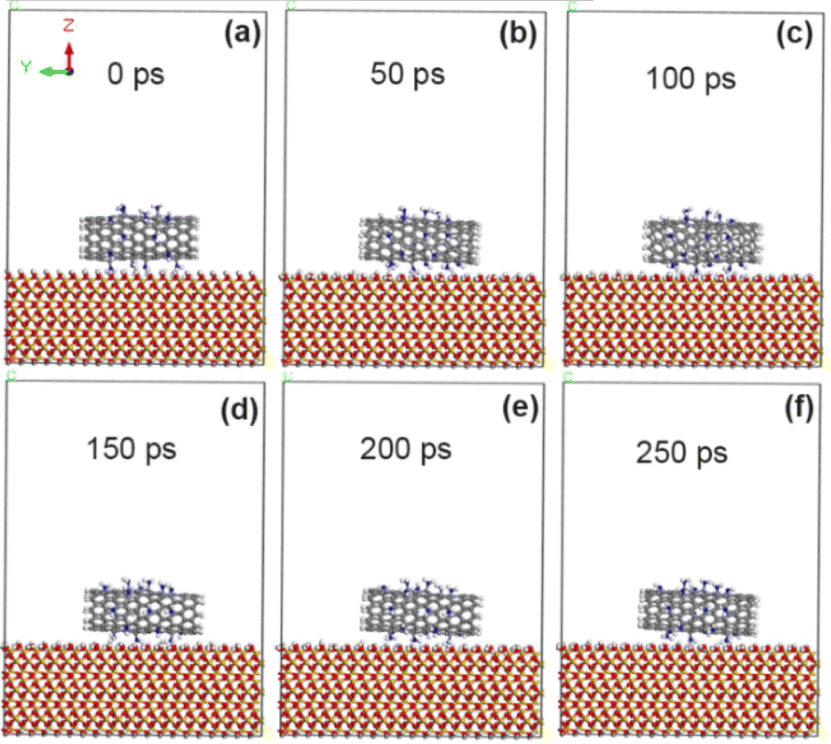
As shown in Fig. 9, CNT is always stably adsorbed on the surface of SiO2 after 10% amino (–NH2) modification. The calculation results in Tables 5 and 6 show that the adsorption energy (−77.6 kcal mol−1) at the interface between CNT and SiO2 is obviously increased with 10% amino grafting amount, but it is slightly lower than that with 5% amino grafting amount. This is because more amino groups increase the distance between CNT and SiO2 interface (3.25 Å), which reduces the non-chemical bonding energy including van der Waals force energy and electrostatic energy. In addition, compared with the calculation results of the previous two sections, it is found that the adsorption energy between unmodified CNT and SiO2 interface modified by F, COOH and NH2 groups increases in turn, and the adhesion enhancement effect of F, COOH and NH2 groups increases in turn. Combined with the experimental data we reported before,20 the results of atomic force microscopy showed that the adhesion of CNT modified by amino group increased from 23.1 nN to 39 nN, which was significantly increased by 69%, while the adhesion of CNT modified by carboxyl group and fluorine group increased by 39%,18,19 and the effect of amino group modification seemed to be more obvious. However, ion collision will occur in the actual experiment process, which will produce defects and active sites on the CNT walls, and may also react with other exposed atoms, thus affecting the adhesion measurement value. Therefore, the theoretical calculation results can only reveal the mechanism of the enhancement trend, which is not completely consistent with the experimental values.
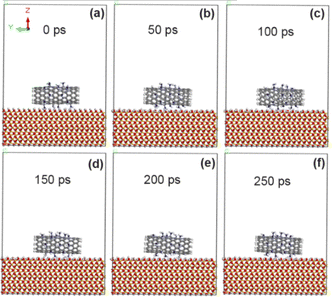 |
| | Fig. 9 State of CNT grafted with 10% amino groups on SiO2 surface at different time. (a) 0 ps; (b) 50 ps; (c) 100 ps; (d) 150 ps; (e) 200 ps; (f) 250 ps. | |
4. Conclusions
In this paper, the previous experimental conclusions were verified by molecular dynamics simulation, and the mechanism of polar group modification on enhancing the adhesion between CNT and SiO2 interface was revealed. Firstly, the simulation results show that the adsorption energy between modified CNT (including F, COOH and NH2 groups) and SiO2 interface is greater than that between unmodified CNT and SiO2 interface, which is consistent with the previous experimental results, further proving that these polar functional groups can enhance the adhesion of CNT. Secondly, the adsorption energy between unmodified CNT and SiO2 interface mainly comes from van der Waals interaction, while the adsorption energy between modified-CNT and SiO2 interface by functional groups comes from the comprehensive action of van der Waals force and electrostatic force. Therefore, it is revealed that the adhesion enhancement of CNT modified by functional groups mainly comes from electrostatic force. Finally, especially for groups with large geometric structure, the van der Waals force decreases slightly. However, in the actual adhesion test process, the shear force measured by CNT modified by groups with large geometric structure and high grafting amount may be stronger, and its surface roughness becomes larger, which hinders the shear movement between sidewalls of CNT and SiO2 interface and increases frictional adhesion. In addition, the influence of CNT walls and defects should be considered in the experiment, and the effect of modification should be analyzed by combining various factors. This work confirmed by theoretical calculation that polar groups can enhance the dry adhesion between carbon nanotubes and the interface, and revealed the adhesion enhancement mechanism. Therefore, when designing carbon nanotube adhesive materials, we can introduce polar groups with more charges through surface modification to enhance the electrostatic interaction at the interface, so as to obtain higher adhesion performance.
Data availability
Data for this article, including [description of data types] are available at [name of repository] at [URL – https://pan.baidu.com/s/1WoG7pk2ZijHeYufX2-n_Fw?pwd=bmum].
Author contributions
Mingyue Lu: writing – review & editing, writing – original draft, methodology, investigation, data curation, funding acquisition. Yanyan Wu: writing – review & editing, data curation. Yang Li: investigation, methodology. Li Ding: writing – review & editing, validation. Zhendong Dai: supervision, funding acquisition. Qinming Gu: software, methodology, validation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (NSFC) (No. 52205311, 52375101) and Technology Project of Changzhou (No. CQ20230074).
References
- P. J. N. Maderson, Nature, 1964, 203, 780–781 CrossRef.
- K. Autumn, M. Sitti and Y. A. Liang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12252–12256 CrossRef CAS PubMed.
- K. Autumn and N. Gravish, Philos. Trans. R. Soc., A, 2008, 366, 1575–1590 CrossRef CAS PubMed.
- S. Gouravaraju, R. A. Sauer and S. S. Gautam, J. Adhes., 2021, 97, 952–983 CrossRef CAS.
- T. E. Higham, M. Zhuang and A. P. Russell, J. Anat., 2021, 239, 1503–1515 CrossRef PubMed.
- L. J. T. Alibardi, Tissue Cell, 2013, 45, 231–240 CrossRef CAS PubMed.
- L. J. P. Alibardi, Protoplasma, 2018, 255, 1785–1797 CrossRef CAS PubMed.
- Y. Song, Z. Wang and Y. Li, Friction, 2022, 10, 44–53 CrossRef.
- S. Sikdar, M. H. Rahman and A. Siddaiah, Robotics, 2022, 11, 143 CrossRef.
- Y. Chen, J. Tian and W. Fu, ACS Appl. Polym. Mater., 2023, 5, 1191–1198 CrossRef CAS.
- W. Duan, Z. Yu and W. Cui, Adv. Colloid Interface Sci., 2023, 313, 102862 CrossRef CAS PubMed.
- Y. Li, H. Zhang and Y. Yao, RSC Adv., 2015, 5, 46749–46759 RSC.
- W. Li, Y. Li and M. Sheng, Langmuir, 2019, 35, 4527–4533 CrossRef CAS PubMed.
- M. Li, M. A. Shakoori and R. Wang, Chin. Phys. Lett., 2024, 41, 016302 CrossRef.
- B. Chen, G. Zhong and P. Goldberg Oppenheimer, ACS Appl. Mater. Interfaces, 2015, 7, 3626–3632 CrossRef CAS PubMed.
- D. Badler, Y. Kligerman and H. J. F. Kasem, Friction, 2023, 11, 1430–1441 CrossRef CAS.
- F. J. M. A. Rajab, MRS Adv., 2023, 8, 343–348 CrossRef CAS.
- M. Lu, Q. He and Y. Li, Carbon, 2019, 142, 592–598 CrossRef CAS.
- M. Lu, Q. He and Y. Li, Nanotechnology, 2020, 31, 345701 CrossRef CAS PubMed.
- M. Lu, Y. Li and X. Zhang, Vacuum, 2023, 216, 112486 CrossRef CAS.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- H. Sun, P. Ren and J. J. C. Fried, Comput. Theor. Polym. Sci., 1998, 8, 229–246 CrossRef CAS.
- J. Xie, Q. Xue and K. Yan, J. Phys. Chem. C, 2009, 113, 14747–14752 CrossRef CAS.
- J. Xie, Q. Xue and H. Chen, J. Phys. Chem. C, 2010, 114, 2100–2107 CrossRef CAS.
- M. Grujicic, G. Cao and W. N. Roy, Appl. Surf. Sci., 2004, 227, 349–363 CrossRef CAS.
- S. Hu, Z. Xia and X. Gao, ACS Appl. Mater. Interfaces, 2012, 4, 1972–1980 CrossRef CAS PubMed.
- L. Qu, L. Dai and M. Stone, Science, 2008, 322, 238–242 CrossRef CAS PubMed.
- P. J. Steinbach and B. R. Brooks, J. Comput. Chem., 1994, 15, 667–683 CrossRef CAS.
- D. A. C. Beck, R. S. Armen and V. Daggett, Biochemistry, 2005, 44, 609–616 CrossRef CAS PubMed.
- A. Ghorai, S. Mahato and S. K. Srivastava, Adv. Funct. Mater., 2022, 32, 2202087 CrossRef CAS.
- E. Gonzalo, M. H. Han and J. M. L. Del Amo, J. Mater. Chem. A, 2014, 2, 18523–18530 RSC.
- T. Nagayasu, C. Yoshioka, K. Imamura and K. Nakanishi, J. Colloid Interface Sci., 2004, 279, 296–306 CrossRef CAS PubMed.
- J. Pan, B. Gao, K. Guo, Y. Gao, X. Xu and Q. Yue, Sci. Total Environ., 2022, 805, 150413 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence