
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen and efficient synthesis of dibenzyl cyanamides and ureas with cyanamide as a block†
Zhongjie Wang a,
Yu Hea,
Fang Wanga,
Yan Wanga,
Hui Luoa,
Jianglong Wu*b and
Jinhui Yang*a
a,
Yu Hea,
Fang Wanga,
Yan Wanga,
Hui Luoa,
Jianglong Wu*b and
Jinhui Yang*a
aState Key Laboratory of High-Efficiency Utilization of Coal and Green Chemical Engineering, School of Chemistry and Chemical Engineering, Ningxia University, Yinchuan 750021, China. E-mail: yang_jh@nxu.edu.cn
bSchool of Chemistry and Chemical Engineering, Ningxia Normal University, Guyuan 756000, China. E-mail: 1548696132@qq.com
First published on 29th July 2024
Abstract
A method for the two-step synthesis of dibenzyl cyanamide and dibenzyl urea via cyanamide is presented. This approach is both efficient and environmentally friendly. Various N,N-dibenzyl ureas could be obtained by reactions of N,N-dibenzyl cyanamides and N,N-dibenzyl cyanamides as intermediates formed from cyanamide. In the absence of metal, ligand and hydrogen peroxide as the oxidant, products with moderate yields have been obtained under mild conditions. Key features include the use of widely available and easily handled cyanamide sources as starting materials.
Cyanation is one of the most important reactions in both organic synthesis and the chemical industry. Nevertheless, these methods often encounter disadvantages, like the production of equal quantities of metal waste, the contamination of the metal catalysts, or the creation of hazardous HCN gas. Consequently, many “non-metallic” organic compounds containing cyano groups have been investigated with the purpose to form carbon-cyano bonds.1,2 There are several methods for synthesizing substituted cyanamide. (1) A common approach is the reaction of amines with halogenated cyanide,3 but these cyanides are highly toxic. (2) Further techniques involve thiourea dehydrosulfurization, hydration of urea, and the transformations from isocyanates, isocyanides, or isothiocyanates.4 (3) Cyanogenic metals to introduce cyanide, such as lithium cyanide.5 (4) Substitution of secondary amines react with cyanide reagents.6
In 1828, the German chemist Friedrich Wohler synthesized urea.7 The capacity of urea and its derivatives to create many stable bonds of hydrogen with proteins and receptor targets makes them crucial in drug discovery and medicinal chemistry. This includes the creation of anticancer, antibacterial, anticonvulsive, anti-HIV, antidiabetic agents, and other medicinal drugs.8 Furthermore, cyanamide is a significant fine chemical feedstock with a unique chemistry characterized by a promiscuous nitrogen–carbon–nitrogen bond. The amino groups' chemistry substituted with nitrile in the cyanamide molecule is characterized by the unique duality of the nucleophilic sp3-amino nitrogen and the electrophilic nitrile unit.9 Cyanamide is therefore a good reagent for cyanation. Further studies on the synthesis of urea from cyanamide derivatives are rarely reported. López group used bromocarbonitriles and dibenzylamines to prepare functionalized cyanamides. They are prepared for functionalized ureas employing the phosphinous acid-based complexes as catalysts, water as a solvent with low metal loading (Scheme 1a and c).10 Xi and colleagues utilized a combination of different synthetic approaches, including both heterogeneous and homogeneous techniques. N-Containing organic compounds can be synthesized via N2 gas and suitable carbon sources, Li2CN2. Many valuable cyanamides, carbodiimides, N-aryl cyanamides, and 1,2,4-triazole derivatives have been synthesized by Li2CN2 (Scheme 1b).5 Qu has reported that the thiolate-bridged diiron compound Me-ligand reacts with various nitriles to give R-ligand, which can realize the hydration of the nitrile ligand under ambient conditions to give ligand. Finally, the amide complex with HBF4·Et2O in the existence of nitriles delivers to the related amides (Scheme 1c).11 Beller revealed a technique employing the iron-catalyzed interaction of urea with a nucleophile. The process of carbamoylation of alcohols enables the production of N-unsubstituted carbamates and its associated products. The use of amines as nucleophiles lead to the corresponding substituted ureas via a selective transamidation reaction (Scheme 1d).12 Our research group is also devoted to the construction of this area.13
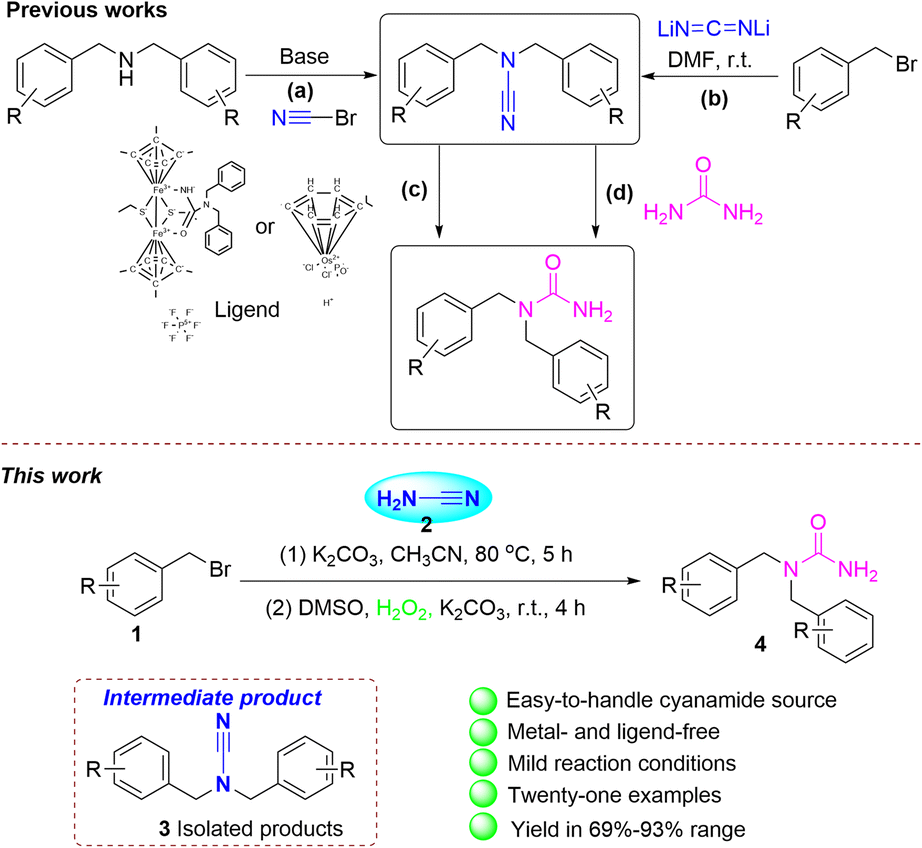 | ||
| Scheme 1 Synthesis of N,N-functionalized cyanamides or ureas. | ||
Herein, we indicate a facile two-step synthesis methodology for N,N-difunctionalized ureas. The cyanamide is reacted with benzyl bromides to form the N,N-difunctionalized cyanamide intermediates. These intermediates are then exposed to mild, open-air, and metal- and ligand-free conditions for reaction with the green oxidant hydrogen peroxide to give N,N-difunctionalized ureas (Scheme 1e). Further investigation revealed that 1,1-bis(2-bromobenzyl) urea underwent a ring closure reaction, resulting in the formation of 3-(2-bromobenzyl)-3,4-dihydroquinazolin-2(1H)-one. The intended product was successfully synthesized on a gram scale with good yield.
As presented in Table 1, we initially initiated our studies by investigating that (bromomethyl)benzene 1a (0.40 mmol), cyanamide 2a (0.20 mmol) and t-BuOK (0.40 mmol) were combined with 5 mL of MeCN solution, stirred and refluxed for 5 h to provide the matching novel amide 3a in 32% yield (entry 1). Having confirmed the feasibility of the synthesis of N,N-difunctionalized cyanamides, we then evaluated different conditions such as temperature, solvent, base, and atmospheres on the reaction of the model. The types of bases were first screened, and it was found that strong bases, whether organic or inorganic, were not conducive to yield improvement (entries 1–4), while potassium carbonate and caesium carbonate were a more excellent performance at 75% and 74% yield (entries 5 and 6). Although the yield using caesium carbonate was almost equal to that using potassium carbonate, we chose to use potassium carbonate as the basis for this experiment because of its raw material source and economic value. Subsequent investigation of solvents showed that acetonitrile (MeCN) was the best-performing solvent, while dichloromethane (DCM), dimethyl sulfoxide (DMSO), tetrahydrofuran (THF), ethyl acetate (EA), dimethyl formamide (DMF), ethyl acetate (EtOH), and methanol (MeOH) gave lower yields than MeCN (entries 7–12). The investigations showed that the effect of solvent on the reaction could achieve relatively good yields in polar non-protonated solvents. In contrast, proton polar solvents had relatively low yields. In addition, the target product was uncaptured in water as the only solvent (entry 13). Subsequently, we investigated the impact of temperature and the quantity of base on the reaction. Our findings revealed that the optimal temperature for the reaction was 80 °C. Moreover, we observed that further increasing the temperature did not lead to any additional rise in the final result of the reaction (entry 14). We focused on the reaction time: if the experiment was run for up to 4 h, the reaction was incomplete; if the reaction was run for up to 5 h, the ingredients disappeared completely; if it was extended to 6 h, the yield would not increase further (entry 15). We also investigated the amount of benzyl bromide added, the amount of alkali added, and the inert gas atmosphere (entries 16–18). Consequently, the reaction circumstances were adjusted as outlined below: (bromomethyl)benzene 1a (0.44 mmol), cyanamide 2a (0.2 mmol), and K2CO3 (0.6 mmol) were mixed in 5 mL of MeCN solution and stirred for 5 h at 80 °C under air atmosphere.
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Temp. (°C) | Solvent (5 mL) | Time (h) | Yield (%) |
| a Conditions of reaction: 1a (0.44 mmol), 2a (0.20 mmol), base (0.60 mmol) in solvent (5 mL) stirred for 5 h at 80 °C under air, isolated yields.b 80 °C and 90 °C.c 4 h or 6 h.d 1a (0.40 mmol, 0.48 mmol).e K2CO3 (0.50 mmol, 0.70 mmol).f Argon atmosphere. | |||||
| 1 | t-BuOK | 80 | MeCN | 5 | 32 |
| 2 | NaOH | 80 | MeCN | 5 | Trace |
| 3 | KOH | 80 | MeCN | 5 | Trace |
| 4 | Na2CO3 | 80 | MeCN | 5 | 60 |
| 5 | K2CO3 | 80 | MeCN | 5 | 75 |
| 6 | Cs2CO3 | 80 | MeCN | 5 | 74 |
| 7 | K2CO3 | 80 | DMSO | 5 | 50 |
| 8 | K2CO3 | 80 | DCM | 5 | 42 |
| 9 | K2CO3 | 80 | THF | 5 | 66 |
| 10 | K2CO3 | 80 | EA | 5 | 61 |
| 11 | K2CO3 | 80 | MeOH | 5 | 36 |
| 12 | K2CO3 | 80 | EtOH | 5 | 32 |
| 13 | K2CO3 | 80 | H2O | 8 | Trace |
| 14b | K2CO3 | 70/90 | MeCN | 5 | 55/72 |
| 15c | K2CO3 | 80 | MeCN | 4/6 | 62/73 |
| 16d | K2CO3 | 80 | MeCN | 5 | 70/73 |
| 17e | K2CO3 | 80 | MeCN | 5 | 64/70 |
| 18f | K2CO3 | 35 | MeCN | 5 | 74 |
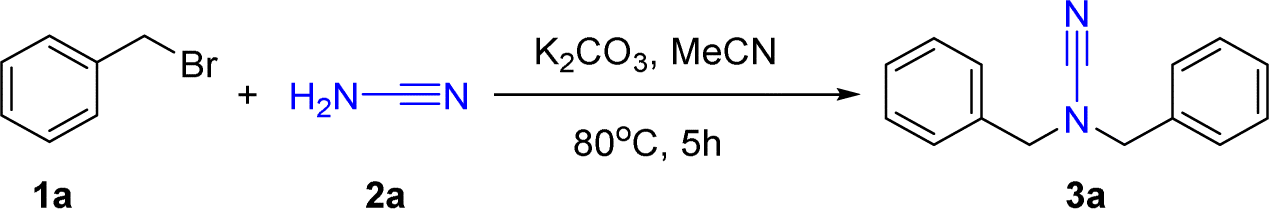
We continued our research on N,N-difunctionalized cyanamides with the hope of using them to synthesise urea derivatives. To enhance the reaction parameters for the sequential creation of rings in 1,1-dibenzylurea 4a, our preliminary examination began with N,N-dibenzyl cyanamide 3a (0.2 mmol) and K2CO3 (0.2 mmol), and an oxidizing agent (0.2 mmol) as substrates of a model in DMSO in an open atmosphere at the temperature of the room. The hydrogen oxide as oxidization, the above reaction did not proceed at all after 5 h (Table 2, entry 1). To induce the desired response, we conducted a model reaction employing H2SO4, NaClO, O2 and H2O2 as oxidizing agents (entries 2–5). The process yielded a novel product, 1,1-dibenzyl-urea (4a), with a 76% yield employing H2O2. The product was separated and analyzed by NMR. Based on the findings mentioned above, the reaction conditions were further optimized by adjusting several factors to enhance its efficiency. Subsequent investigation of the solvents revealed that DMSO yield was the best at 76%, whereas DMF, MeCN, THF, DMC, EA, ethanol (EtOH), and methanol (MeOH) resulted in lower yields (entries 6–11). After optimizing the solvent, we investigated hydrogen peroxide or base in the absence of organic solvent conditions, but these did not react (entries 12 and 13). In order to enhance the reaction's effectiveness, we proceeded to evaluate larger amounts of H2O2. It was discovered that using 1.2 equivalents of H2O2 resulted in the outcome we wanted 4a with an 85% yield (entry 14). However, raising the quantity of hydrogen peroxide beyond this point did not cause any further improvement in the yield. The temperature of the reaction is a crucial factor in oxidation processes; thus, we ultimately examined the impact of temperature. The test reaction was conducted throughout a temperature range of room temperature to 40 °C, and no significant alteration in the reaction's conclusion was detected (entry 15). The experiment was detected by TLC; it was found that the raw material pad disappeared completely in about 4 h, indicating that the reaction was complete (entry 16). Furthermore, we conducted investigations on our technique in the presence of an inert argon atmosphere; nevertheless, it did not enhance the end result yield (entry 17). Subsequently, our results confirm that the optimum conditions of the reaction consist of N,N-dibenzyl cyanamide 3a (0.20 mmol), K2CO3 (0.2 mmol), and hydrogen peroxide (0.24 mmol, 30% a.q.) in DMSO (3 mL) solvent for 5 h in an open atmosphere at room temperature.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Oxidation | Base | Temp. (°C) | Solvent (mL) | Time (h) | Yield (%) |
| a Conditions of reaction: 3a (0.20 mmol), oxidizing agent (0.24 mmol, 30% a.q.), K2CO3 (0.20 mmol) in solvent (3 mL) stirred for 4 h at r.t. under air, isolated yields.b Oxidizing agent (0.24 mmol, 0.30 mmol, 30% a.q.).c Temperature 30 °C, 40 °C.d Time 4 h, 6 h.e Argon atmosphere. | ||||||
| 1 | H2O | K2CO3 | r.t. | DMSO | 5 | Nr |
| 2 | O2 | K2CO3 | r.t. | DMSO | 5 | Nr |
| 3 | NaClO (a.q.) | K2CO3 | r.t. | DMSO | 5 | 26 |
| 4 | H2SO4 | K2CO3 | r.t. | DMSO | 5 | 42 |
| 5 | H2O2 (a.q.) | K2CO3 | r.t. | DMSO | 5 | 76 |
| 6 | H2O2 (a.q.) | K2CO3 | r.t. | MeCN | 5 | 22 |
| 7 | H2O2 (a.q.) | K2CO3 | r.t. | THF | 5 | 35 |
| 8 | H2O2 (a.q.) | K2CO3 | r.t. | MeOH | 5 | 24 |
| 9 | H2O2 (a.q.) | K2CO3 | r.t. | EtOH | 5 | 30 |
| 10 | H2O2 (a.q.) | K2CO3 | r.t. | EA | 5 | 28 |
| 11 | H2O2 (a.q.) | K2CO3 | r.t. | DCM | 5 | 23 |
| 12 | H2O2 (a.q.) | K2CO3 | r.t. | — | 5 | Nr |
| 13 | H2O2 (a.q.) | — | r.t. | DMSO | 5 | Trace |
| 14b | H2O2 (a.q.) | K2CO3 | r.t. | DMSO | 5 | 85/84 |
| 15c | H2O2 (a.q.) | K2CO3 | 30/40 | DMSO | 5 | 80/81 |
| 16d | H2O2 (a.q.) | K2CO3 | r.t. | DMSO | 4/6 | 75/85 |
| 17e | H2O2 (a.q.) | K2CO3 | r.t. | DMSO | 5 | 85 |
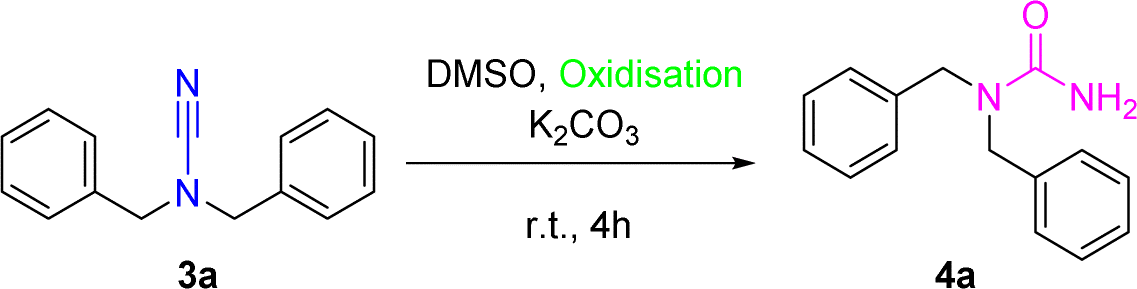
The ideal conditions, as illustrated in Scheme 2, allowed for the successful formation of a wide variety of products from benzyl bromide substrates with different functional groups. Particularly, the various substituted benzyl bromides (R = H, CH3, CF3, CH3O, OCF3, NO2, t-Bu, F, Cl, Br, and I) instead of 1a afforded products 3a–3t in moderate to high yields (52–87%), and the electron-donating substituents (1h, 1j, 1p, yields 78–87%) exhibited more favorable effects than the electron-withdrawing substituents (1e, 1i, 1s, yields 50–80%). However, the presence of easily eliminated substituents such as iodine caused a significant reduction in the yield of products 3n (yield 62%). Subsequent investigations showed that the incorporation of a chloro group at the ortho-(1c), meta-(1g), or para-(1l) positions resulted in the formation of the target products 3c, 3g, and 3l in moderate to high yields (65%, 78%, and 86%). Additionally, the reactions exhibited a broad tolerance for para-derivatives resulting in high yields of the corresponding products with electron-donating groups (80–87%, 3o–3r) and electron-withdrawing groups (62–86%, 3k–3n, 3s–3t). Furthermore, the structure of 3m and 3s are verified by X-ray analysis (CCDC: 2324482, 2324486).
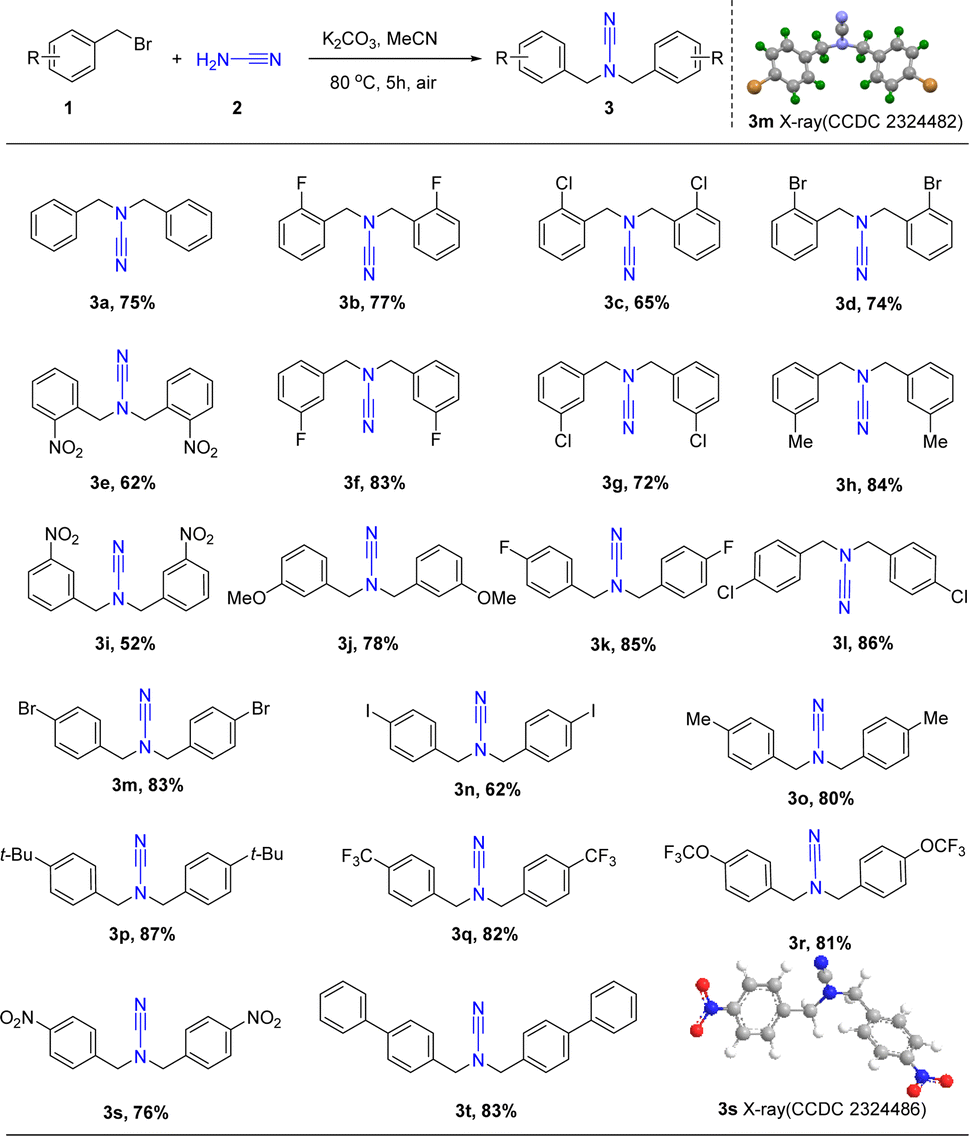 | ||
| Scheme 2 The substrate scope of the N,N-functionalized cyanamide 3. Conditions of reaction: 1a (0.44 mmol), 2a (0.20 mmol), K2CO3 (0.60 mmol) in MeCN (5 mL) stirred for 5 h at 80 °C at air, isolated yields. | ||
However, functionalized cyanamide was not our ultimate goal; we synthesized functionalized ureas from functionalized cyanamides under metal- and ligand-free, green oxidant hydrogen peroxide conditions. According to the ideal circumstances, the significance of the reactions of different functionalized cyanamide derivatives 3 with hydrogen peroxide to produce a number of N,N-functionalized urea 4 was examined by using K2CO3 as a base in DMSO for 5 h at room temperature (Scheme 3). The reactions exhibited a high degree of substrate tolerance. The study also examined the reactions of several ortho-substituted functionalized cyanamides with halogens (F, Cl, Br, and NO2) and hydrogen peroxide. These reactions yielded compounds with yields ranging from 65% to 78% (4b–4e). X-ray analysis was used to elucidate the structures of the products, which were confirmed to be 4b (CCDC: 2324490). When the conditions were optimized, the reaction utilized the meta-substituted substrates 4f and 4j, resulting in yields of 68–76% for each substrate, respectively. Following that, the substitution impacts on the amide ring that arises from the para-position were evaluated. The study revealed that the reactions were able to withstand a broad spectrum of cyanamide derivatives that were functionalized at the para-position. This resulted in the production of the associated compounds with high yields ranging from 79% to 88%. Notably, the reactions were successful with both electron-withdrawing groups (4k–4n, 4s–4t) and electron-donating groups (4o–4r). Although the nitro-functionalized cyanamides in the ortho-, meta- or para-positions obtained the corresponding functionalized urea by the reaction, the yields were lower than the other substituents. In comparison with the electron-withdrawing-substituted cyanamides, the findings showed that the electron-donating-substituted cyanamides were more advantageous to the synthesis of the intended product.
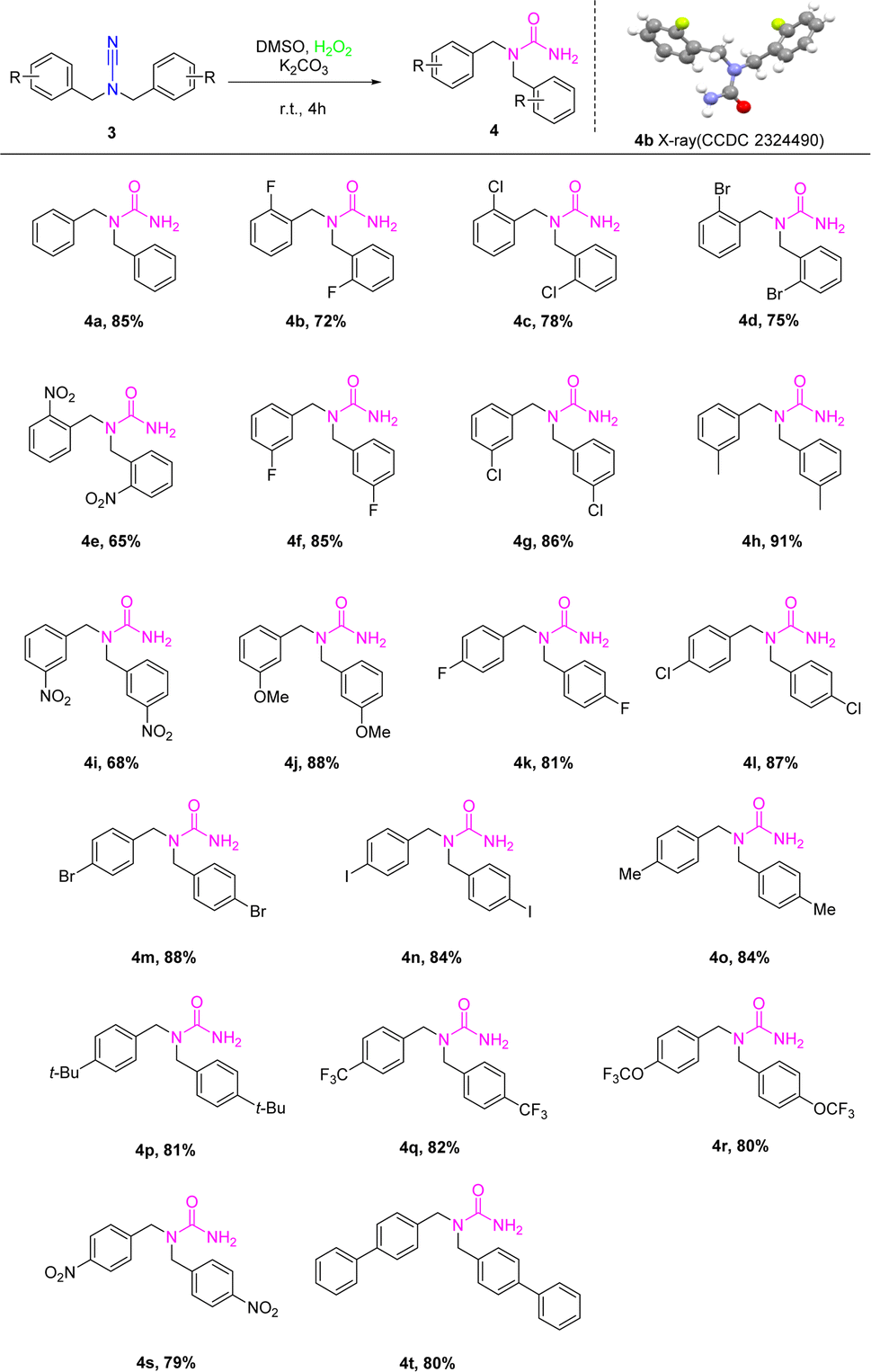 | ||
| Scheme 3 Substrate scope of the N,N-functionalized urea 4. Reaction conditions: 3a (0.20 mmol), H2O2 (0.24 mmol, 30% a.q.), K2CO3 (0.20 mmol) in DMSO (3 mL) stirred for 4 h at r.t. under air, isolated yields. | ||
The synthesis of 1,1-dibenzyl urea 4a by reaction of 1a with cyanamide could also be performed on a gram scale. The reaction of 3.73 g of 1a and 0.42 g of cyanamide 2a in 25 mL of MeCN was conducted at standard conditions to provide 1.63 g of 4a in 84% separated yield (Scheme 4). The findings indicated that the optimal circumstances for that reaction were well-suited for large-scale activities.
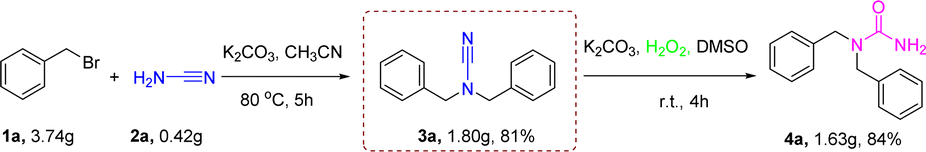 | ||
| Scheme 4 Gram-scale synthesis of 4a. Conditions of reaction: 1a (22.0 mmol), 2a (10.0 mmol), K2CO3 (30.0 mmol) in MeCN (25 mL) stirred for 5 h at 80 °C under air atmosphere, isolated yields; 3a (8.1 mmol), hydrogen peroxide (9.7 mmol), K2CO3 (8.1 mmol) in DMSO (15 mL) stirred for 4 h at r.t. under air, isolated yields. | ||
To our surprise, we synthesized 3-(2-bromobenzyl)-3,4-dihydroquinazolin-2(1H)-one (5a) using 1,1-bis(2-bromobenzyl)urea (4d, 0.20 mmol) as raw material, cuprous iodide (0.02 mmol) as catalyst and potassium tert-butoxide (0.60 mol) as base in DMSO solvent (2 mL) at 130 °C for 8 h in air atmosphere (Scheme 5). It provided a new technique for the creation of 3-(2-bromobenzyl)-3,4-dihydroquinazolin-2(1H)-one (51 mg, 80% yield).
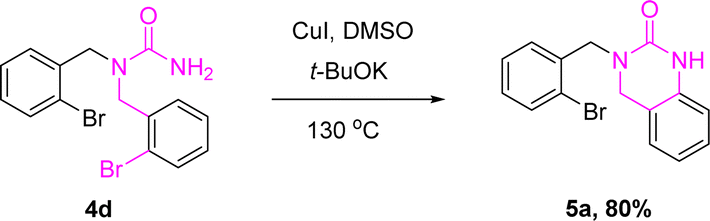 | ||
| Scheme 5 Synthesis of derivatives 5a. Reaction conditions: 3d (0.2 mmol), CuI (0.02 mmol), t-BuOK (0.6 mmol) in DMSO (2 mL) stirred for 8 h at 130 °C under air, isolated yields. | ||
Based on our control experimentation and the literatures,5,11,12 we have suggested a viable method for the production of compound 3–5 (Scheme 6). Initially, cyanamide 2a loses hydrogen to form the nitrogen anion in the existence of potassium carbonate, with concomitant removal of the bromine atom by benzyl bromides to form the carbon-positive ion and the attack of the nitrogen anion on the carbon-positive ion to form the functionalized cyanamides A. The functionalized cyanamide A continues to shed the hydrogen atom of the amino group under alkaline conditions to form a nitrogen-negative ion, which then attacks the carbon-positive ion of benzyl bromide to form N,N-difunctionalized cyanamide 3. The carbon–nitrogen triple bond in compound 3 undergoes an oxidative addition reaction with peroxide ion under alkaline conditions to form intermediate B.14 Intermediate B removes hydrogen ions under the action of potassium carbonate to form intermediate C. The C oxidizes the solvent (DMSO) to dimethyl sulfone, which is hydrolyzed and isomerized to form intermediate D, finally giving. Product 4d is complexed with cuprous iodide and, based on a strong base (t-BuOK) action, a hydrogen atom is removed from the urea by a cyclization reaction to give 5a.
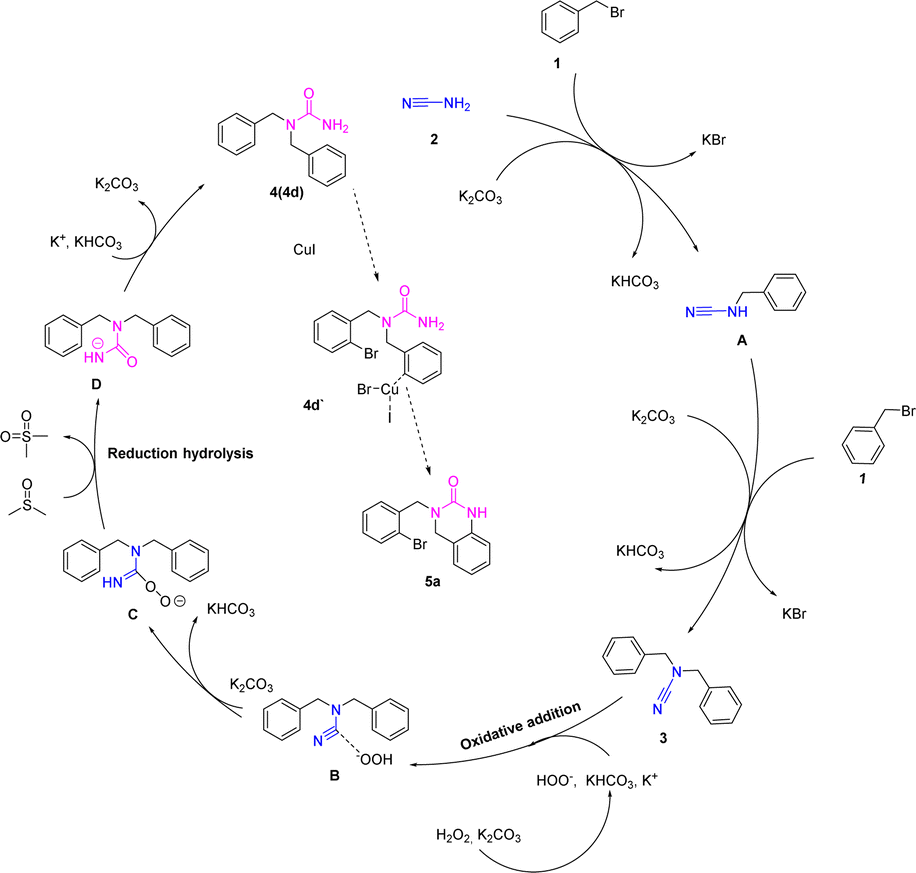 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusions
An environmentally friendly and two-step synthesis for N,N-functionalized cyanamides and ureas method has been developed using cyanamide as the building block. The low toxicity, high safety, and easy handling of cyanamide and benzyl bromide form substituted cyanamides, and the substituted cyanamides are further oxidized to N,N-functionalized ureas using green hydrogen peroxide oxidant under mild conditions and metal- and ligand-free. The successful execution of the gram-scale experiments indicated the feasibility of the reaction pathway for large-scale production.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Key Research and Development Project of Ningxia (2021BEG02001, 2021BEE03003), National Natural Science Foundation of China (Grant No. 21861031, 21362025), Ningxia Science and Technology Leading Talents (2019, 2022), Institute of Local Cooperation Project of Chinese Academy of Engineering (2023NXZX1), Ningxia Natural Science Foundation (2022AAC03047).References
- (a) J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948–11959 CrossRef CAS PubMed; (b) G. Yan, Y. Zhang and J. Wang, Adv. Synth. Catal., 2017, 359, 4068–4105 CrossRef CAS.
- (a) H. Tian, C. Y. Ding, R. Z. Liao, M. Li and C. Tang, J. Am. Chem. Soc., 2024, 6, 11801–11810 CrossRef PubMed; (b) Y. Ping, Q. Ding and Y. Peng, Catal, 2016, 6, 5989–6005 CAS; (c) D. Zhao, P. Xu and T. Ritter, Chem, 2019, 5, 97–107 CrossRef CAS; (d) S. Shi and M. Szostak, Org. Lett., 2017, 19, 3095–3098 CrossRef CAS PubMed; (e) C. B. Kelly, K. M. Lambert, M. A. J. M. Ovian, W. F. Bailey and N. E. Leadbeater, Angew. Chem., Int. Ed., 2015, 54, 4241–4245 CrossRef CAS PubMed; (f) Y. Yan, J. Sun, G. Li, L. Yang, W. Zhang, R. Cao, C. Wang, J. Xiao and D. Xue, Org. Lett., 2022, 24, 2271–2275 CrossRef CAS PubMed; (g) H. Li, J. Chen, J. Dong and W. Kong, Org. Lett., 2021, 23, 6466–6470 CrossRef CAS PubMed; (h) Z. J. Qi, C. N. Hu, Y. W. Zhong, C. Cai and G. P. Lu, Org. Chem. Front., 2021, 8, 3137–3149 RSC; (i) Y. Zhu, M. Zhao, W. Lu, L. Li and Z. Shen, Org. Lett., 2015, 17, 2602–2605 CrossRef CAS PubMed; (j) M. Chaitanya and P. Anbarasan, Org. Lett., 2015, 17, 3766–3769 CrossRef CAS PubMed.
- (a) C. Zhu, J.-B. Xia and C. Chen, Org. Lett., 2014, 16, 247–249 CrossRef CAS PubMed; (b) I. Furusawa, T. Suzuki, Y. Yu and T. Arai, Adv. Synth. Catal., 2023, 365, 3247–3252 CrossRef CAS.
- (a) T. J. Gong, B. Xiao, W.-M. Cheng, W. Su, J. Xu, Z. J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 10630–10633 CrossRef CAS PubMed; (b) M. Chaitanya, D. Yadagiri and P. Anbarasan, Org. Lett., 2013, 15, 4960–4963 CrossRef CAS PubMed; (c) Y. Yang, Y. Zhang and J. Wang, Org. Lett., 2011, 13, 5608–5611 CrossRef CAS PubMed; (d) P. Anbarasan, H. Neumann and M. Beller, Chem.–Eur. J., 2011, 17, 4217–4222 CrossRef CAS PubMed; (e) P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 519–522 CrossRef CAS PubMed; (f) E. Duchamp and S. Hanessian, Org. Lett., 2020, 22, 8487–8491 CrossRef CAS PubMed.
- L. J. Wu, Q. Wang, J. Guo, J. Wei, P. Chen and Z. Xi, Angew. Chem., Int. Ed., 2023, 135, e202219298 CrossRef.
- (a) G. Talavera, J. Pena and M. Alcarazo, J. Am. Chem. Soc., 2015, 137, 8704–8707 CrossRef CAS PubMed; (b) J. N. Ayres, K. B. Ling and L. C. Morrill, Org. Lett., 2016, 18, 5528–5531 CrossRef CAS PubMed; (c) F. Teng, J. T. Yu, Y. Jiang, H. Yang and J. Cheng, Chem. Commun., 2014, 50, 8412–8415 RSC; (d) F. Teng, J. T. Yu, Z. Zhou, H. Chu and J. Cheng, J. Org. Chem., 2015, 80, 2822–2826 CrossRef CAS PubMed; (e) P. Kumari, R. Nagpal, P. Chauhan, V. Yatindranath and S. Chauhan, J. Chem. Sci., 2015, 127, 13–18 CrossRef CAS; (f) C. C. Lin, T. H. Hsieh, P. Y. Liao, Z. Y. Liao, C. W. Chang, Y. C. Shih, W. H. Yeh and T. C. Chien, Org. Lett., 2014, 16, 892–895 CrossRef CAS PubMed; (g) N. Kuhl, S. Raval and R. D. Cohen, Org. Lett., 2019, 21, 1268–1272 CrossRef CAS PubMed.
- (a) F. Wöhler, Ann. Phys. Chem., 1828, 37, 330–333 Search PubMed; (b) P. J. Ramberg, Ambix, 2000, 47, 170–195 CrossRef CAS PubMed.
- (a) A. K. Ghosh and M. Brindisi, J. Med. Chem., 2019, 63, 2751–2788 CrossRef PubMed; (b) I. Gallou, Org. Prep. Proced. Int., 2007, 39, 355–383 CrossRef CAS; (c) J. Regan, S. Breitfelder, P. Cirillo, T. Gilmore, A. G. Graham, E. Hickey, B. Klaus, J. Madwed, M. Moriak, N. Moss, C. Pargellis, S. Pav, A. Proto, A. Swinamer, L. Tong and C. Torcellini, J. Med. Chem., 2002, 45, 2994–3008 CrossRef CAS PubMed.
- (a) M. R Prabhath, L. Williams, S. V. Bhat and P. Sharma, Molecules, 2017, 22, 615 CrossRef PubMed; (b) D. Yang, Y. Wang, H. Yang, T. Liu and H. Fu, Adv. Synth. Catal., 2012, 354, 477–482 CrossRef CAS; (c) N. Mishra, A. S. Singh, A. K. Agrahari, S. K. Singh, M. Singh and V. K. Tiwari, ACS. Comb. Sci., 2019, 21, 389–399 CrossRef CAS PubMed.
- R. González-Fernández, D. Álvarez, P. Crochet, V. Cadierno, M. I. Menéndez and R. López, Catal. Sci. Technol., 2020, 10, 4084–4098 RSC.
- P. Tong, D. W. Yang, Y. Li, B. M. Wang and J. P. Qu, Organometallics, 2015, 34, 3571–3576 CrossRef CAS.
- M. Peña-López, H. Neumann and M. Beller, ChemSusChem, 2016, 9, 2233–2238 CrossRef PubMed.
- (a) J. L. Wu, Y. F. Ma, Y. Wang, C. Y. Wang, H. Luo, D. J. Li and J. H. Yang, Green Chem., 2023, 25, 3425–3430 RSC; (b) J. L. Wu, Z. J. Wang, C. Y. Wang, Y. Wang, H. J. Li, H. Luo, H. Li, F. Q. Wang, D. J. Li and J. H. Yang, Chin. J. Org. Chem., 2023, 43, 436–454 CrossRef CAS.
- S. Zhang, G. Li, L. Li, X. Deng, G. Zhao, X. Cui and Z. Tang, ACS Catal., 2019, 10, 245–252 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2324482, 2324486 and 2324490. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra04286g |
| This journal is © The Royal Society of Chemistry 2024 |