
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First-principles study of phase transition and the structural, energetic and electronic properties of pristine and transition metal (Fe/Co/Ti)-doped layered MoS2 as anode materials for sodium-ion batteries†
Wenlong
Xi
 and
Patrick H.-L.
Sit
*
and
Patrick H.-L.
Sit
*
School of Energy and Environment, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China. E-mail: patrick.h.sit@cityu.edu.hk
First published on 9th February 2025
Abstract
In this work, we apply first-principles density functional theory (DFT) calculations to study the intercalation of Na atoms into the pristine and transition metal (TM)-doped MoS2 (MxMo1−xS2) layers. Our results show that TM atom doping enhances the binding of the Na atoms between the MxMo1−xS2 (M: Fe/Co/Ti) layers. Moreover, we find that Na intercalation facilitates the transition from the 2H phase to the 1T phase of MoS2 in agreement with previous findings. However, Fe and Co doping is found to promote such transition; conversely, Ti doping is found to delay this transition. MxMo1−xS2 have metallic properties, and the doping increases the average open-circuit voltage (OCV) of the 1T and 2H phase MxMo1−xS2. This work provides a new perspective on the phase change mechanism of transition metal dichalcogenides and valuable theoretical insights for the development of doped MoS2 nanomaterials in Na-ion battery applications.
1. Introduction
In recent years, the demand for advanced energy storage systems has been rapidly increasing, driven by the need for efficient and sustainable energy solutions. Among various energy storage technologies, rechargeable batteries have emerged as a key player in powering portable electronics,1 electric vehicles,2 and grid-scale energy storage applications.3 Lithium-ion batteries (LIBs) have dominated the market due to their high energy density and long cycle life.4–6 However, the limited availability and high cost of lithium resources have prompted researchers to explore alternative battery materials.7One such alternative is sodium-ion batteries (NIBs), which utilize sodium ions instead of lithium ions for energy storage.8 Sodium, being more abundant and widely distributed than lithium, offers a potential solution to the resource limitations associated with LIBs. NIBs have the potential to deliver comparable energy storage performance while offering a more sustainable and cost-effective alternative.9
The performance of a battery is closely related to the properties of its electrode materials. In the case of NIBs, the development of high-performance anode materials is crucial to achieve the desired energy storage capabilities.10 While in LIBs the commonly used anode material for LIBs is graphite,11 the Na atoms cannot electrochemically intercalate into graphite because of the weak Na–C intercalation, and sodium graphite intercalation compounds (Na-GICs) are thermodynamically unstable.12,13 Therefore, there has been an urgent need in the exploration and optimization of anode materials for NIBs.
Transition metal dichalcogenides (TMDs) have attracted considerable attention as potential anode materials for NIBs.14–16 They are also potentially promising electrode materials for batteries, such as Li+,17 K+,18 and Mg2+.19 TMDs are a class of layered compounds consisting of transition metal atoms sandwiched between chalcogen (sulfur, selenium, or tellurium) atoms. These materials exhibit unique structural, electronic, and electrochemical properties that make them promising candidates for energy storage applications. Neighboring layers of MoS2 are attracted together by feeble van der Waals forces, and there exists sufficient room between the layers to accommodate alkali metal ions during the charging process.20
MoS2 is a typical TMD that shows great potential as an anode material for NIBs.21 MoS2 mainly has two phases, 1T and 2H phases. Previous work shows that during the continuous intercalation of sodium atoms, MoS2 undergoes phase transition from the 2H phase to the 1T phase.21,22 Some researchers have conducted first-principles simulation studies on the intercalation process of Na atoms in MoS2. Mortazavi et al. examined the efficacy of the layered MoS2 as a host electrode material for NIBs and found that the strong binding of Na with MoS2 can prohibit Na cluster formation.22 Wang et al. studied the edge engineering of MoS2 nanoribbons as a high-performance electrode material for Na-ion batteries and found that the Mo edge was half saturated by S, which helps stabilize the MoS2 nanoribbons as well as offer more intercalation sites for Na.23
On the other hand, incorporating heteroatoms through doping represents a promising strategy for augmenting the electronic, magnetic, and structural properties of materials.24 Transition metal (such as Fe and Co) doping can adjust the electronic properties of MoS2 to enhance the energy storage and conversion performance of MoS2 in batteries.25,26 Moreover, there have been a number of studies on Fe, Co, and Ti-doped MoS2 as battery anode materials, and it was found that such doping can result in different advantages like more efficient charge transfer or increase in the cation diffusion rate.27–30 However, the understanding of the microscopic details of the intercalation processes and the impact of transition metal doping on the electrochemical performance of MoS2 is still limited.
In this work, we employ first-principles density functional theory (DFT) calculations to investigate the intercalation process of sodium atoms within the pristine and transition metal (TM)-doped MoS2 layers. We explore the binding energy of sodium atoms in the interlayer region of TM-doped MoS2 and investigate the influence of TM doping on the intercalation behavior. This study aims to provide an in-depth understanding of the electrochemical properties and phase transition mechanisms in TM-doped MoS2, thus contributing to the development of high-performance anode materials for NIBs.
2. Computational method
In this study, we employed the PWscf module of the quantum ESPRESSO (QE) package to perform the density functional theory (DFT) calculations.31,32 The UltraSoft pseudopotential (USPP) was utilized.33 To account for the van der Waals (vdW) interactions, we incorporated semiempirical Grimme's DFT-D2,34,35 in conjunction with the Perdew–Burke–Ernzerhof (PBE) exchange correlation functional.36 The kinetic energy cutoff for the wavefunctions was set as 60 Ry, and the kinetic energy cutoff for the augmented charge density was set as 480 Ry. In all the geometry optimization, the variable cell optimization was used, and the Brillouin zone was sampled using a 6 × 6 × 3 k-point grid. For the MoS2 model, a 2 × 2 × 2 periodic supercell was used, so that there are two MoS2 monolayers in the periodic supercell. The optimized lattice parameters of the pristine 1T and 2H phase MoS2 are shown in Table S1 (ESI†).3. Results and discussion
3.1. Na binding sites and intercalation structures in the pristine and TM-doped MoS2
There are two possible Na atom binding sites in both the 1T and 2H phases, namely, the octahedral site (Oh) in which the Na binds to the six nearest sulfur atoms, and the tetrahedral site (Td) in which the Na binds to the four nearest sulfur atoms, as displayed in Fig. 1(a)–(d). Fig. 1(e) and (f) show the doped structure of MoS2, where M is the transition metal dopant: Fe/Co/Ti. The TM-doped MoS2 structure was constructed by replacing one Mo atom in the bi-layer pristine MoS2 supercell structure with a transition metal atom, so that there are 1 M atom, 7 Mo atoms and 16 S atoms in each simulation. Here, we denote the TM-doped MoS2 structure as M0.125Mo0.875S2, where the M is the doped metal atom Fe/Co/Ti in the TM-doped structure. We considered eight different Na atom intercalation concentrations, namely NaxM0.125Mo0.875S2, (x = 0.125, 0.250, 0.375, 0.500, 0.625, 0.750, 0.875 and 1.000). | ||
| Fig. 1 Octahedral binding sites of Na in the (a) 1T and (b) 2H phase MoS2, tetrahedral binding sites of Na in the (c) 1T and (d) 2H phase MoS2, and the top view of TM-doped multi-layered (e) 1T and (f) 2H phase MoS2. | ||
We first examined the binding sites and binding energies of single Na atom intercalation in the pristine layered MoS2. We found that the octahedral site is a more stable binding site having a binding energy around 0.2 eV lower than that of the tetrahedral site, which is similar to previous works.22,37 Then, we studied the intercalation of a single Na atom in M0.125Mo0.875S2, and the Oh site was still found to be more stable. We also found that as shown in Fig. S1 (ESI†), for the Ti-doped MoS2 in the 1T phase, the energy of the case with Na intercalated near the Ti is slightly lower than that of Na further away from the Ti by 0.05 eV. For the Ti-doped MoS2 in the 2H phase, it is also energetically more favorable for the Na close to the Ti by 0.04 eV. On the contrary, for the Fe-doped MoS2 in the 1T phase, the energy of the structure of Na intercalated further away from the Fe is 0.12 eV lower than the case of Na near the Fe. For the 2H phase, the case for Na further from the Fe is more energetically favorable by 0.09 eV. For the Co-doped MoS2, the 1T phase and 2H phase favor Na further from the dopant by 0.10 eV and 0.26 eV, respectively. Therefore, if the Na atom is far away from the Co or Fe dopant, the energy is lower. On the contrary, the Na atom tends to be close to the Ti dopant.
Next, we studied the intercalation of different concentrations of Na atoms into the 1T and 2H phases of the pristine and TM-doped MoS2 supercell structure. Since the energy of the Na atom at the octahedral sites is more stable than that at the tetrahedral sites, we considered the configurations of each component on the premise of intercalating the Na to the octahedral sites first. In all configurations, the Na atoms were initially distributed in octahedral positions as far away from each other as possible to minimize their interaction and avoid congregation. For each configuration in Fig. S2 (ESI†), the structure was fully optimized, and the configuration with the lowest energy was selected for analysis. The formula for calculating the binding energy of Na atoms in layered M0.125Mo0.875S2 is as follows:
| Eb(x) = E(NaxM0.125Mo0.875S2) − E(M0.125Mo0.875S2) − xE(Na) | (1) |
Fig. 2 shows the Na binding energy over the entire composition range for various stably bound structures of NaxM0.125Mo0.875S2 in the 1T and 2H phases. Overall, as the Na concentration increases, the binding energy gradually becomes less negative, which is mainly due to the increased electrostatic repulsion between the Na at a higher concentration and the weakened interaction between the Na and the layered M0.125Mo0.875S2.41 For the 1T pristine structure, we found that when the Na concentration is 0.125, the binding energy per atom is less negative than when the Na concentration is 0.25. We will discuss the possible reason in Section 3.2.
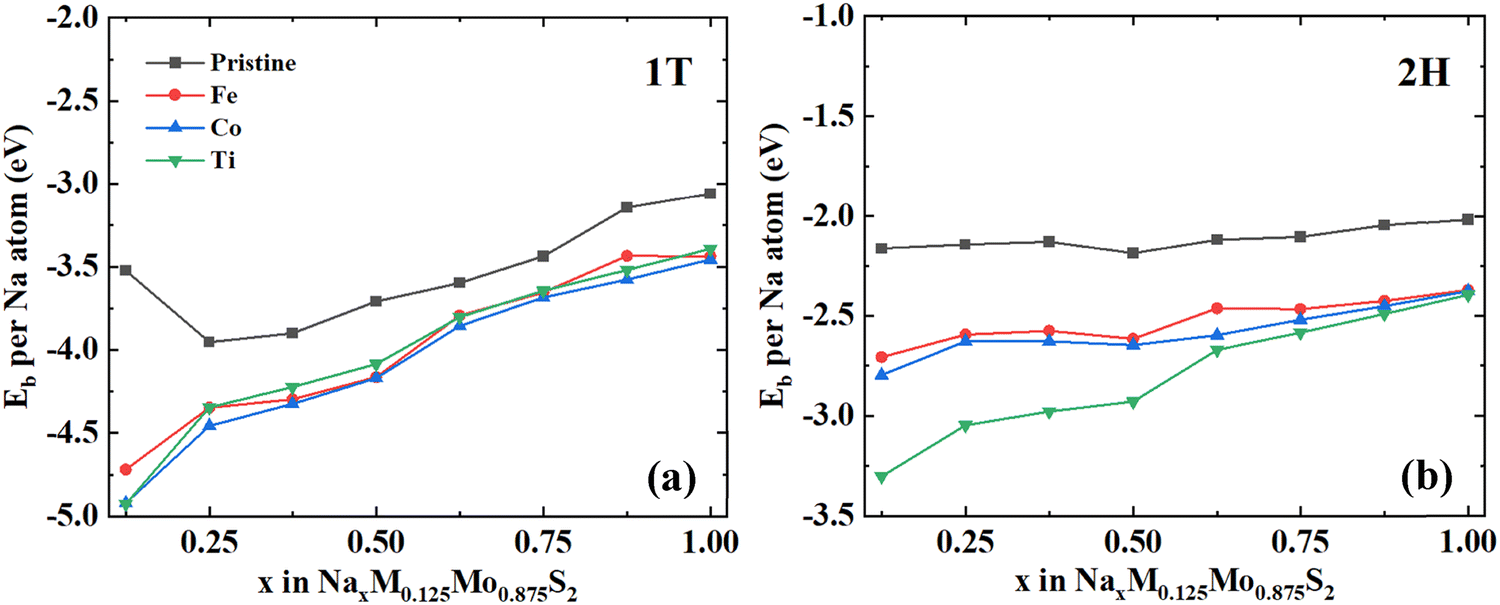 | ||
| Fig. 2 Binding energy per Na atom for the (a) 1T phase and (b) 2H phase of the pristine MoS2 and M0.125Mo0.875S2, M: Fe/Co/Ti. | ||
It is obvious that the binding energy of all structures in the 1T phase of NaxM0.125Mo0.875S2 is more negative than that of the 2H phase, usually 1–2 eV lower than that of the 2H phase. Through comparison, we can find that, for both the 1T phase and the 2H phase, Fe, Co, and Ti doping will strengthen the binding of the Na with the layered structure in the entire composition range. It shows that doping is more beneficial to the combination of Na atoms and M0.125Mo0.875S2. Interestingly, for the doped structures with Fe, Co, and Ti in the 1T phase, the binding energies are similar. For the 2H phase in Fig. 2b, the binding energies of the Na atoms in the Fe- and Co-doped structures are similar. However, the binding energy of the Ti-doped structure is always lower than that of Fe and Co until the Na concentration is 0.875. After that, the binding energy of the Ti-doped structure becomes similar to that in the Fe- and Co-doped structure. From Fig. S3 (ESI†) we can know that in the range of x = 0–0.25, interlayer spacing will increase rapidly, and then it will fluctuate in a narrow range. Fe/Co/Ti doping will reduce the interlayer spacing around 0.1–0.3 Å, compared with the pristine structure.
3.2. Phase transition of the layered pristine MoS2 and M0.125Mo0.875S2 upon Na intercalation
As it was found that the lithiation or sodiation of the TMD materials will cause the transition from the 2H to the 1T phase compound,21,42 we next studied the relative stability of the pristine MoS2 and M0.125Mo0.875S2 upon Na intercalation. Since 2H-MoS2 and 2H-M0.125Mo0.875S2 are more stable phases, respectively, we define the formation energy of all NaxM0.125Mo0.875S2 intercalation compounds with respect to 2H-M0.125Mo0.875S2 and the Na body-centered cubic crystal as the reference state as:| Ef(x) = E(NaxM0.125Mo0.875S2) − E(2H-M0.125Mo0.875S2) − xE(Nabcc) | (2) |
In Fig. 3a, the formation energy per atom of the 1T pristine structure is around 3 eV when the Na concentration is 0.125, and the structure is much less stable than the 2H phase at this time. After doping with the Fe, Co, and Ti elements, the formation energy of the layered NaxM0.125Mo0.875S2 compound decreases. For the 1T phase shown in Fig. 3b–d, especially when the Na concentration is 0.125, although the formation energy is still positive, it is reduced by more than 50%. When the Na concentration is 0.25, the formation energy is reduced to a negative value. For the pristine MoS2 structure, the formation energy decreases to a negative value only when the third Na atom is intercalated.
 | ||
| Fig. 3 Formation energy per Na atom for 1T and 2H phases of (a) pristine, (b) Fe0.125Mo0.875S2, (c) Co0.125Mo0.875S2, and (d) Ti0.125Mo0.875S2. | ||
It shows that the doping of Fe, Co, and Ti will promote the intercalation of Na atoms into the interlayers of the 1T phase of the M0.125Mo0.875S2 compound. Moreover, the transition points of the phase change from the 2H phase to the 1T phase (the concentration of Na atoms when the phase change occurs) were found to be Pristine: 0.365, Fe-doped: 0.355, Co-doped: 0.311, and Ti-doped: 0.503. In the process of continuous intercalation of sodium atoms, Fe and Co doping will accelerate the transition from the 2H phase to the 1T phase, which is consistent with previous experimental studies;28,43 conversely, Ti doping will delay this transition. In order to further confirm the effect of doping on phase transition, we increased the number of dopant atoms. As shown in Fig. S4 (ESI†), the phase change transition points of NaxFe0.25Mo0.75S2, NaxCo0.25Mo0.75S2, and NaxTi0.25Mo0.75S2 are 0.309, 0.171 and 0.698, respectively. Doping with more Fe/Co atoms can lead to an even earlier phase transition, while the opposite is true for Ti.
3.3. Bader charge analysis
To further study the charge transfer during the intercalation process in pristine MoS2 and M0.125Mo0.875S2, we performed the Bader charge analysis. Through this analysis, we can gain insights into the nature of the interactions and the distribution of electrons in the system. Fig. 4 shows charges of different atoms in the intercalated systems at different intercalation concentrations of Na. For the 1T phase, in the average Bader charges of the Na atoms and Mo atoms both show a downward trend as shown in Fig. 4a and b. The trends of the changes of the Na charge are very similar irrespective of the existence or the type of doping. Moreover, the decreasing charge can be related to the fact that the MoS2 host cannot effectively hold additional electrons when more Na intercalate. On the other hand, the charge of the dopant fluctuates within a certain range without an obvious upward or downward trend, as shown in Fig. 4c. Interestingly, the charges of Fe and Co are both lower than that of Mo, while the charge of Ti is higher than that of Mo, as shown in Fig. 4b. Compared with Mo, Fe and Co donate less electrons. Under the same Na atom intercalation concentration, the S charge of the Fe/Co doping structure is less negative than that of the pristine structure. At this time, the entire system has a stronger ability to receive electrons from the Na atoms, while Ti donates more electrons to the system, causing the charge of S to become more negative as shown in Fig. 4d, forming a stronger ionic bond between S and the metal atoms. The average charge of S shows a steady downward trend with increasing Na concentration. | ||
| Fig. 4 Bader charge for (a) Na, (b) Mo, (c) M and (d) S in the 1T phase, and (e) Na, (f) Mo, (g) M and (h) S in the 2H phase of pristine MoS2 and M0.125Mo0.875S2, M: Fe/Co/Ti and the black lines in (c) and (g) are the average charge of Mo in pristine MoS2. | ||
For the 2H phase, the average charge of the Na atoms decreases faster than that of the 1T phase, and the average charge of the Na atoms varies greatly in different doped structures as shown in Fig. 4e. This shows that in the 2H phase, Na atoms provide more electrons to MoS2 to form a stable structure compared with the 1T phase. Compared with Fig. 4a, the charge of Na in the 1T phase is larger than that in the 2H phase when the Na concentration is 1, which means the 1T MoS2 can still host more electrons than the 2H phase when more Na atoms intercalate. The average charge of the Mo atoms shows an overall downward trend, some fluctuates from one concentration to the next as shown in Fig. 4f. In the 2H phase, the charges of Fe and Co are lower than that of Mo, while the charge of Ti is higher than that of Mo, but the charge of Ti constantly decreases, and the charges of Fe and Co show an upward trend as in Fig. 4g. The average charge change of S is basically consistent with that of the 1T phase as shown in Fig. 4h.
In order to further analyze the impact of doping on 1T and 2H phases, we calculated the number of electrons in the d-orbitals of the TM ions in each structure. As shown in Fig. 5a, the number of d-orbital electrons in the 1T phase initially decreases and then increases as the intercalation concentration increases. For Fe and Co doping, the number of electrons begin to increase when the Na concentration is 0.25. For Ti doping, when the Na concentration is 0.375, the number of electrons in the Mo d-orbital begins to increase. Such transition points seem to be correlated with the critical points for phase transitions. Also, d-orbital electrons in the 2H phase show an upward trend as seen in Fig. 5b.
 | ||
| Fig. 5 Mo d orbital electrons of (a) 1T and (b) 2H phase pristine MoS2 and M0.125Mo0.875S2, M: Fe/Co/Ti. | ||
Moreover, we found that the Mo–S distance (dMo–S) of the 1T phase is generally larger than that of the 2H phase as shown in Table S1 (ESI†), which explains why the number of d-orbital electrons in the 1T phase is smaller than that of the 2H phase due to the smaller sharing of the electrons in S to the d-orbitals of the Mo in the 1T phase. From Table S1 (ESI†), compared with dMo–S, the M–S distance (dM–S) is smaller for the 1T and 2H phases in the Fe/Co/Ti-doped structure. For the Fe/Co doped structures, the dM–S (2.31–2.32 Å) in the 1T phase is smaller than in 2H (2.34 Å), and it is significantly smaller than dMo–S (2.42–2.44 Å) of the pristine structure; while for the Ti-doped structure, the dM–S in the 1T phase is still larger than in 2H, and they are only slightly smaller than the dMo–S of the pristine structure. The dM–S in Fe/Co-doped structures decreases to 2.31–2.34 Å compared to the dMo–S in pristine MoS2, and there is a stronger interaction between the Fe/Co atoms and the S atoms.
3.4. Open-circuit voltages (OCVs)
Next, we explore the battery voltage of the pristine MoS2 and the three doped M0.125Mo0.875S2 compounds as the anode materials. First, we compare the relative stabilities of these configurations to determine the more stable configuration formed during the intercalation of Na atoms. The relative formation energy for a given NaxMoS2 composition can be calculated as:44| Erf(x) = E(NaxMoS2) − xE(NaMoS2) − (1−x)E(MoS2) | (3) |
NaMoS2 and MoS2 refer to the fully sodiated and desodiumized phases, respectively. The convex hull of the formation energy of all these structures was constructed as a function of the Na concentration (x). Thermodynamically stable phases are identified by energy convex hulls. The convex hull plots are plots of formation energy with respect to the composition that connects phases that are lower in energy than any other phases or a linear combination of phases at that composition. Phases that lie on the convex hull line are thermodynamically stable and the ones above the lines are either metastable or unstable. All identified stable phases are based on static first principles calculations corresponding to the 0 K ground state, as are the calculated voltages. Using this formula to obtain the corresponding convex hull and determine the corresponding thermodynamically stable phase, we obtained the results shown in Fig. 6.
 | ||
| Fig. 6 Convex hull of the 1T and 2H phase (a) pristine, (b) Fe, (c) Co, and (d) Ti doping MoS2 structures. | ||
For each concentration (x) of the NaxM0.125Mo0.875S2 compound, the OCV (V) relative to Na/Na+ is calculated as follows:45
 | (4) |
Fig. 7 Shows the OCV curves of the pristine and Fe/Co/Ti-doped MoS2 with respect to the concentration of the Na in the intercalation compounds. We found that after doping with the Fe, Co, and Ti elements, the initial OCV of the 1T phase increased from 2.6 V to 3.5–3.8 V, and the initial OCV of the 2H phase increased from 0.8 V to 1.4–2 V. As the concentration of intercalated Na atoms increases, the OCV is in a downward trend. With the continuous intercalation of Na atoms, the intercalation of additional Na atoms into the layered MoS2 and M0.125Mo0.875S2 compounds becomes more difficult. This is because the electrostatic repulsion between alkali ions increases, resulting in weaker binding of alkali ions.46
 | ||
| Fig. 7 OCV curves of 1T and 2H phase (a) pristine, (b) Fe, (c) Co, and (d) Ti doping MoS2 structures. | ||
Since the formation of Na0.125M0.125Mo0.875S2 and Na0.125MoS2 is thermodynamically unstable, we will ignore these compounds when calculating the average OCV. The average OCV of the final 1T phase pristine MoS2 and Fe/Co/Ti-doped MoS2 are 1.20 V, 1.77 V, 1.76 V and 1.71 V, respectively. The average OCVs of 2H phase pristine MoS2 and Fe/Co/Ti-doped MoS2 are 0.65 V, 0.98 V, 1.05 V and 1.03 V, respectively, and the pristine results are consistent with some other experimental results.21,47 Therefore, doping with Fe, Co, and Ti, the average OCV increases around 45% and 50% for the 1T and 2H phases, and M0.125Mo0.875S2 is suitable as sodium-ion battery electrode materials.
3.5. Electronic properties
The electronic conductivity of the electrode materials is a key aspect that determines the battery performance, as highly conducting materials provide a low internal resistance to the current flow. Therefore, we calculated the projected density of states (PDOS) to further elucidate the electronic conduction behavior of the pristine MoS2 and doped M0.125Mo0.875S2 compounds after intercalation of the Na atom. Previous studies have shown that the pristine 1T phase MoS2 is metallic, while the 2H phase MoS2 is a semiconductor. The projected density of states (PDOS) can be used to show the electronic states and interactions of different atoms in a material. For the doped structures, similar to defective structures, if one atom is replaced, the electronic structure of the surrounding atoms may be rearranged, causing the intensity and position of some hybrid peaks to change.48After Fe, Co and Ti doping, the electronic states of M0.125Mo0.875S2 at the Fermi level are non-zero. The 1T phase continues to maintain the metallic properties as shown in Fig. 8a–d, and the electronic states near the Fermi level are mainly contributed by Mo and S atoms. The contribution of dopant atoms to the Fermi level is smaller, but when the metal dopants are introduced into the 1T phase, they lead to significant alterations in the electronic structure by introducing additional states within the bandgap or shifting the existing bands, while the 2H phase changes from semiconductor properties with a well-defined bandgap to metallic properties as shown in Fig. 8e–h. But the main valence and conduction bands remain relatively stable upon metal doping, which means that the overall electronic structure does not change as drastically as in the 1T phase. After Fe, Co and Ti doping, the electronic states around the Fermi level are enhanced and then increase changer transfer and conductivity of MoS2. Similar findings that doping to the 1T phase leads to larger changes in the PDOS than that to the 2H phase were also reported in previous studies.49–51 The layered M0.125Mo0.875S2 compound can retain the metallic properties during the Na atom intercalation process, as shown in Fig. 8i–p, proving the feasibility of M0.125Mo0.875S2 as an anode material for sodium-ion batteries.
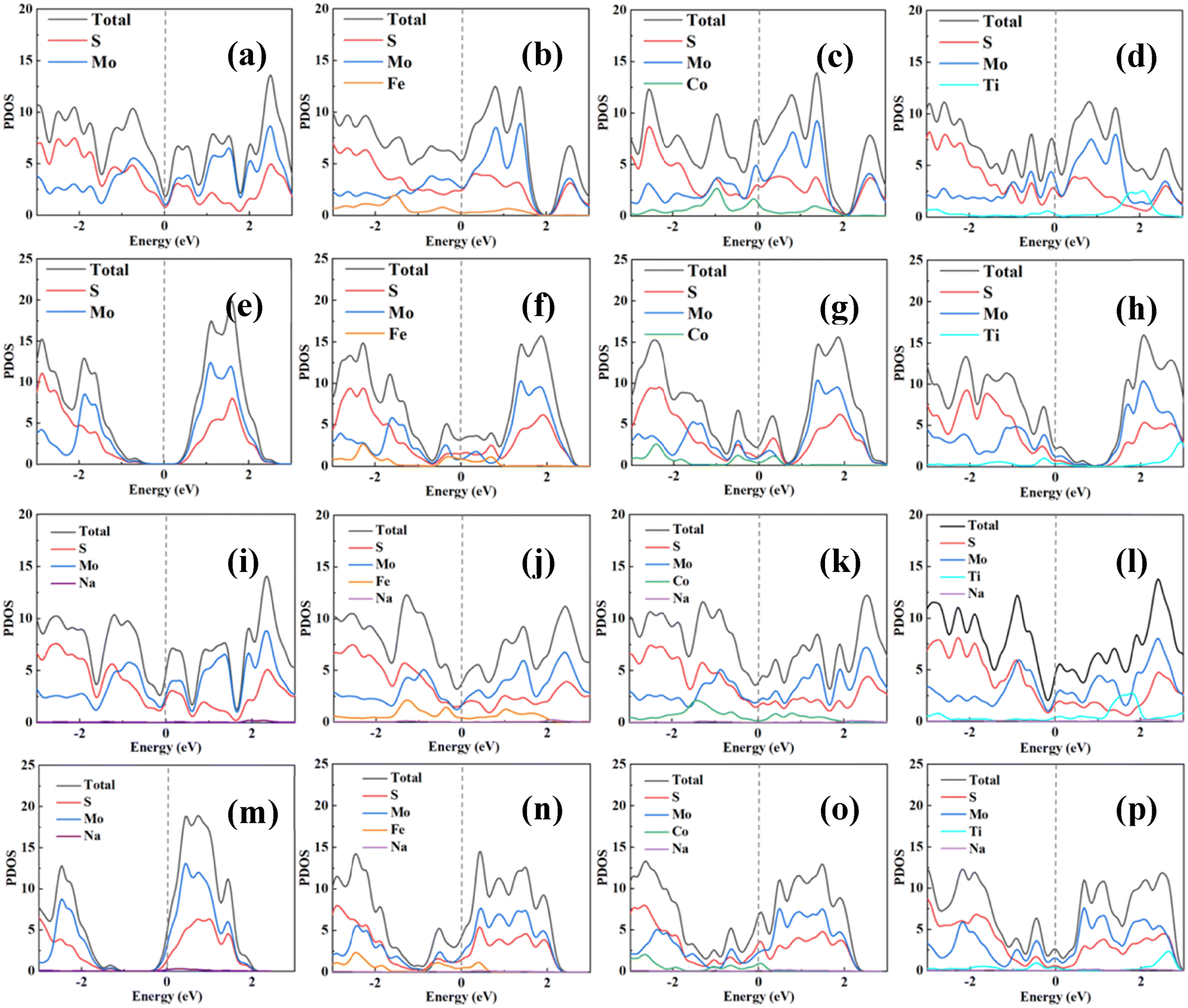 | ||
| Fig. 8 Projected density of states for (a) pristine, (b) Fe-doped, (c) Co-doped and (d) Ti-doped 1T phase MoS2 and (e) pristine, (f) Fe-doped, (g) Co-doped and (h) Ti-doped 2H phase MoS2. Na atom intercalation in (i) pristine, (j) Fe-doped, (k) Co-doped and (l) Ti-doped 1T phase MoS2 and Na atom intercalation in (m) pristine, (n) Fe-doped, (o) Co-doped, and (p) Ti-doped 2H phase MoS2. The Fermi level is set to zero eV. | ||
4. Conclusions
In summary, the intercalation process of Na atoms between the pristine and transition metal-doped MoS2 layers was studied by DFT calculations. Firstly, the binding energy of Na atoms between doped MoS2 layers was found to be more negative than the pristine structure and promotes the intercalation of Na atoms between layers. Secondly, Fe and Co doping to the MoS2 was found to facilitate the transition from the 2H phase to the 1T phase of the MoS2. Conversely, Ti doping will delay this transition. Thirdly, after doping with Fe, Co, and Ti, the average OCV increases around 45% and 50% for the 1T and 2H phase, respectively. Finally, the 1T and 2H phase M0.125Mo0.875S2 all have metallic properties and embody ideal Na storage and conductive properties when used as the sodium-ion battery anode material.Author contributions
Wenlong Xi: conducted the investigation, experiments and data analysis, and wrote the manuscript. Patrick Sit: involved in the discussions and supervised, bore the idea, obtained the research resources, managed the project, and supervised the research, reviewed & edited the manuscript.Data availability
Data will be made available on request.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
We acknowledge the support from the CityU SRG Funds (7005723) and the Research Matching Grant Scheme (RMGS - PJ9229008) of the Hong Kong Special Administrative Region, China. This work was carried out using the computational facilities, CityU Burgundy, managed and provided by the Computing Services Centre at the City University of Hong Kong (https://www.cityu.edu.hk/) and the computer facilities managed by the School of Energy and Environment of the City University of Hong Kong.References
- Y. Liang, C.-Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J.-Q. Huang, D. Yu, Y. Liu, M.-M. Titirici, Y.-L. Chueh, H. Yu and Q. Zhang, InfoMat, 2019, 1, 6–32 CrossRef CAS
.
- Y. Li, J. Yang and J. Song, Renewable Sustainable Energy Rev., 2017, 74, 19–25 CrossRef
- Z. Zhu, T. Jiang, M. Ali, Y. Meng, Y. Jin, Y. Cui and W. Chen, Chem. Rev., 2022, 122, 16610–16751 CrossRef CAS PubMed
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed
- G. Zhou, F. Li and H. M. Cheng, 2014.
- L. Peng, Y. Zhu, D. Chen, R. S. Ruoff and G. Yu, Adv. Energy Mater., 2016, 6, 1600025 CrossRef
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed
- J. Cui, S. Yao and J.-K. Kim, Energy Storage Mater., 2017, 7, 64–114 CrossRef
- M. Mamoor, Y. Li, L. Wang, Z. Jing, B. Wang, G. Qu, L. Kong, Y. Li, Z. Guo and L. Xu, Green Energy Resources, 2023, 1, 100033 CrossRef
- H. Zhang, Y. Yang, D. Ren, L. Wang and X. He, Energy Storage Mater., 2021, 36, 147–170 CrossRef
- D. P. DiVincenzo and E. J. Mele, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 2538–2553 CrossRef CAS PubMed
- O. Lenchuk, P. Adelhelm and D. Mollenhauer, Phys. Chem. Chem. Phys., 2019, 21, 19378–19390 RSC
- Q. Yun, L. Li, Z. Hu, Q. Lu, B. Chen and H. Zhang, Adv. Mater., 2020, 32, 1903826 CrossRef CAS PubMed
- H. Tan, Y. Feng, X. Rui, Y. Yu and S. Huang, Small Methods, 2020, 4, 1900563 CrossRef CAS
- X. Yuan, Z. Chen, B. Huang, Y. He and N. Zhou, J. Phys. Chem. C, 2021, 125, 10226–10234 CrossRef CAS
- L. Wang, Z. Xu, W. Wang and X. Bai, J. Am. Chem. Soc., 2014, 136, 6693–6697 CrossRef CAS PubMed
- J. Rehman, X. Fan, A. Laref, V. A. Dinh and W. T. Zheng, J. Alloys Compd., 2021, 865, 158782 CrossRef CAS
- Y. Liang, H. D. Yoo, Y. Li, J. Shuai, H. A. Calderon, F. C. Robles Hernandez, L. C. Grabow and Y. Yao, Nano Lett., 2015, 15, 2194–2202 CrossRef CAS PubMed
- Y. Zhang, L. Zhang, T. A. Lv, P. K. Chu and K. Huo, ChemSusChem, 2020, 13, 1114–1154 CrossRef CAS PubMed
- Q. Li, Z. Yao, J. Wu, S. Mitra, S. Hao, T. S. Sahu, Y. Li, C. Wolverton and V. P. Dravid, Nano Energy, 2017, 38, 342–349 CrossRef CAS
- M. Mortazavi, C. Wang, J. Deng, V. B. Shenoy and N. V. Medhekar, J. Power Sources, 2014, 268, 279–286 CrossRef CAS
- J. Wang, Q. Zhao and J. Chen, Chin. J. Chem., 2017, 35, 896–902 CrossRef CAS
- L. Guo, J. Li, H. Wang, N. Zhao, C. Shi, L. Ma, C. He, F. He and E. Liu, Phys. Rev. Appl., 2018, 9, 024010 CrossRef CAS
- K. Qi, Z. Yuan, Y. Hou, R. Zhao and B. Zhang, Appl. Surf. Sci., 2019, 483, 688–695 CrossRef CAS
- W. Liu, C. Luo, S. Zhang, B. Zhang, J. Ma, X. Wang, W. Liu, Z. Li, Q. H. Yang and W. Lv, ACS Nano, 2021, 15, 7491–7499 CrossRef CAS PubMed
- I. Williamson, S. Li, A. Correa Hernandez, M. Lawson, Y. Chen and L. Li, Chem. Phys. Lett., 2017, 674, 157–163 CrossRef CAS
- G. Dou, L. He, L. Huang, D. Wang, Y. Wang, M. Guo and G. Zhang, J. Energy Storage, 2024, 98, 113060 CrossRef
- X. Yao, J. Zhu, H. Wang, K. Yang, Y. Shu and J. He, Appl. Surf. Sci., 2022, 587, 152842 CrossRef CAS
- S. Xu, H. Chen, C. Li, R. Nie, Y. Yang, M. Zhou, X. Zhang and H. Zhou, J. Alloys Compd., 2023, 962, 171199 CrossRef CAS
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H. Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H. V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- V. Barone, M. Casarin, D. Forrer, M. Pavone, M. Sambi and A. Vittadini, J. Comput. Chem., 2009, 30, 934–939 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- J. Shuai, H. D. Yoo, Y. Liang, Y. Li, Y. Yao and L. C. Grabow, Mater. Res. Express, 2016, 3, 064001 CrossRef
- J. Hu, B. Xu, S. A. Yang, S. Guan, C. Ouyang and Y. Yao, ACS Appl. Mater. Interfaces, 2015, 7, 24016–24022 CrossRef CAS PubMed
- D. Wang, Y. Gao, Y. Liu, D. Jin, Y. Gogotsi, X. Meng, F. Du, G. Chen and Y. Wei, J. Phys. Chem. C, 2017, 121, 13025–13034 CrossRef CAS
- H. Liu, Y. Cai, Z. Guo and J. Zhou, ACS Omega, 2022, 7, 17756–17764 CrossRef CAS PubMed
- R. Zhao, C. Wan, P. Qian and X. Ju, Surf. Interfaces, 2023, 38, 102851 CrossRef CAS
- S. Fan, X. Zou, H. Du, L. Gan, C. Xu, W. Lv, Y.-B. He, Q.-H. Yang, F. Kang and J. Li, J. Phys. Chem. C, 2017, 121, 13599–13605 CrossRef CAS
- B. Gao, Y. Zhao, X. Du, D. Li, S. Ding, Y. Li, C. Xiao and Z. Song, Chem. Eng. J., 2021, 411, 128567 CrossRef CAS
- Q. Bai, L. Yang, H. Chen and Y. Mo, Adv. Energy Mater., 2018, 8, 1702998 CrossRef
- M. K. Aydinol, A. F. Kohan and G. Ceder, J. Power Sources, 1997, 68, 664–668 CrossRef CAS
- A. K. Nair, C. M. Da Silva and C. H. Amon, J. Appl. Phys., 2023, 133, 064302 CrossRef CAS
- J. Park, J.-S. Kim, J.-W. Park, T.-H. Nam, K.-W. Kim, J.-H. Ahn, G. Wang and H.-J. Ahn, Electrochim. Acta, 2013, 92, 427–432 CrossRef CAS
- Q. Zhou, Z. Fu, C. Wang, Y. Tang, H. Zhang, L. Yuan and X. Yang, Appl. Surf. Sci., 2015, 356, 1025–1031 CrossRef CAS
- Q. Wang, Y. Wang, Z. Li and X. Ma, Int. J. Hydrogen Energy, 2024, 82, 416–427 CrossRef CAS
- L. Lin, J. Huang, W. Yu, L. Zhu, H. Tao, P. Wang and Y. Guo, Solid State Commun., 2019, 301, 113702 CrossRef CAS
- H. Wei, Y. Gui, J. Kang, W. Wang and C. Tang, Nanomaterials, 2018, 8, 646 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00286a |
| This journal is © the Owner Societies 2025 |