
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudying the effect of dimensions and spacer ligands on the optical properties of 2D lead iodide perovskites†
Orly
Abarbanel
,
Rawan
Hirzalla
,
Leehie
Aridor
 ,
Elisheva
Michman
and
Ido
Hadar
*
,
Elisheva
Michman
and
Ido
Hadar
*
Institute of Chemistry, The Center for Nanoscience and Nanotechnology, The Casali Center for Applied Chemistry, The Hebrew University of Jerusalem, Jerusalem, Israel. E-mail: ido.hadar@mail.huji.ac.il
First published on 13th February 2025
Abstract
In recent years, metal–halide perovskites (MHPs) have emerged as highly promising optoelectronic materials based on their exceptional properties and versatility in applications such as solar cells, light-emitting devices, and radiation detectors. This study investigates the optical properties of two-dimensional (2D) MHPs, with the Ruddlesden–Popper structure, comparing three morphologies–bulk poly-crystals, colloidal nanoplatelets (NPs), and thin films, aiming to bridge between the bulk and nano dimensionalities. By synthesizing bulk 2D MHPs using long alkyl ammonium spacers, typically found in colloidal systems, and NPs using shorter ligands suitable for bulk growth, we elucidate the relationship between these materials’ structural modifications and optical characteristics. We propose the existence of two regions in these 2D MHPs, which differ in their optoelectronic properties and are associated with “bulk” and “surface” regions. Specifically, for poly-crystals, we observe the appearance of a lower energy “bulk” phase associated with the stacking of many 2D sheets, apparent both in absorption and photoluminescence. For NPs, this stacking is hindered, and hence, only the “surface” phase exists. With the elongation of the spacer chain, the poly-crystal becomes more similar to the NPs. For thin films, an interesting phenomenon is observed – the rapid film formation mechanism forces a more colloid-like structure for the shorter ligands and a more poly-crystal-like structure for the longer ones. Overall, this study bridging the different dimensions of 2D MHPs may support new possibilities for future research and development in this innovative field.
 Ido Hadar | Ido Hadar is an assistant professor in the Institute of Chemistry at the Hebrew University of Jerusalem, Israel. He received his Ph.D. (2016) from Hebrew University under the guidance of Prof. Uri Banin, studying the effects of dimensionality on the optical properties of semiconductor nanocrystals. He then joined the group of Prof. Mercouri Kanatzidis at Northwestern University (2016–2020) as a post-doctoral researcher studying semiconductor materials for energy conversion, light emission, and radiation detection. Dr Hadar's lab studies novel soft semiconductor materials with specific interests in structure–property correlations and structural dynamics in the excited state. |
Introduction
Metal–Halide Perovskites (MHPs), a novel family of hybrid organic–inorganic semiconductors, have arisen in the past decade as promising optoelectronic materials suitable for versatile applications.1–15 Initially, MHPs were utilized for solar energy harvesting as thin film photovoltaic devices with high power conversion efficiency, high absorption cross-sections, and efficient charge separation.1–5,8,16–18 In addition, MHPs are fabricated by simple solution processing enabling low-cost and scalable production.8–10,19–22 Structurally, MHPs are based on a connected network of metal–halide octahedra (MX6, M – Pb+2, Sn+2, X – I−, Br−, Cl−). For three-dimensional MHPs, this network is stabilized by a small cation (A – methylammonium, formamidinium, or Cs+),13,23,24 forming a 3D network of corner-sharing octahedra with AMX3 stoichiometry.12,24 The rich and agile chemistry of MHPs, enabled by modifications to the metal,25 halide,1,6,8 and A sites,13 has brought them to the spotlight of materials chemistry research. Such chemical modifications enabled tuning of the optoelectronic properties of MHPs (e.g., band gap) and led to the emergence of additional applications, including light-emitting devices (LEDs), lasers, and high-energy radiation detectors.5–7,12,15,19,26,27Dimensionality reduction is an additional knob for tuning the MHP structure. The dimensionality of the octahedral network can be modified by introducing a bulkier A site cation (e.g., alkylamine chains).13,24,28 2D MHPs formed by terminating the octahedral network in one direction while maintaining corner-sharing connectivity in two dimensions are a typical example.14,22,29–31 The structure of 2D MHPs can be tuned by modifying the bulkier cation, affecting both the crystal structure of the 2D layer (e.g., nearly cubic or heavily distorted) and the stacking of multiple 2D layers.13 By mixing small and bulky cations, quasi-2D MHPs are realized and can form a homologous series with a defined thickness,13,19,32 allowing the bridging between the 2D and 3D MHPs. Most 2D MHP compounds share a few desired properties, such as quantum- and dielectric-confinement, leading to a higher band gap and binding energies, both beneficial for LED applications5,33–36 and scintillation-based detectors.19,37–39 An additional benefit of 2D MHPs is their improved stability under ambient conditions,40 contributing to device longevity both as pure 2D and as mixed 2D–3D geometries.23,32,41–45 All of these have led to vast research on 2D and quasi-2D MHPs and the discovery of many compounds with well-defined structures and properties.
In parallel to the development of bulk MHPs, the research on colloidal MHP nanocrystals (NCs) has thrived as an alternative route to optimize and tune these compounds’ characteristics. The first examples of MHP NCs were based on 3D MHPs with nanometer dimensions.46–48 These NCs showed bright emission with high quantum efficiency without special surface treatments or passivation.24,48 The color of these NCs could be tuned by the halide composition, including alloying.49–53 However, the quantum confinement in these NCs was fairly small.51,54–56 Colloidal 2D MHP nanoplatelets (NPs) combine the desired properties of 3D MHP NCs with the strong quantum confinement that arises from dimensionality reduction.36,48,57–61 The colloidal NPs seem to form 2D layers with similar structures to the bulk 2D and quasi-2D MHPs.13,62–64 Yet, their exact crystal structures are still not as well resolved. Similarly, many modifications of the optical properties have been realized for 2D MHP NPs, but the detailed structure–property correlation in NPs is lacking.
The current research aims to bridge between bulk and nano 2D MHPs and identify the similarities and differences between these regions – from bulk compounds through thin films to colloidal NPs. We chose to focus our research on single-layer 2D MHPs of the Ruddlesden–Popper structure – (A′)2PbI4 (A′ – an alkylammonium spacer with different chain lengths). Generally, short spacers are used for the growth of single or poly-crystals (e.g., butylamine, hexylamine), whereas longer spacers (e.g., octadecylamine) are more common in colloidal NP systems. Even though there are exceptions, this is the general approach due to synthetic limitations and colloidal stabilization aspects. The synthetic route also differs between morphologies. While the bulk material is usually synthesized through a slow, controlled crystallization process,13,14,19,22,29,30,65 colloidal synthesis occurs through fast nucleation and limited growth.13,57,66–70 To achieve our goal, we chose to synthesize bulk 2D MHPs with long spacers usually used for colloids and to synthesize colloid NPs using short ligands suitable for bulk poly-crystals. We studied how the structure and optical properties of these 2D compounds are changed as a function of spacer length and synthetic methods.
Results and discussion
Synthesis
The standard procedure for synthesizing bulk 2D MHPs is through precipitation and crystallization from a saturated halide acid solution (HI).13,19,29,65,71 This is achieved by heating the HI with a suitable Pb precursor (e.g., PbO) to 180–230 °C, until it is fully dissolved. At these temperatures, the spacers are added slowly, and the solution is vigorously stirred until it is clear (an indication of full dissolution). Then, the solution is slowly cooled until precipitation of 2D MHPs begins, followed by slow growth of poly-crystals. This method relies on the solubility gap between the precursors and the 2D MHP in a heated halide acid. The slow cooling induces controlled precipitation and, as a result, larger crystals. For the alkyl-ammonium spacers used in this research, we observed that with each addition of carbons to the alkyl chain, the solubility decreased, and the required temperature for dissolution was increased – this is expected due to the lower polarity of the longer chains. Nevertheless, for alkyl-ammonium spacers with chain lengths of 4 to 10 carbons (butylamine to decylamine, C4–C10), this method worked well enough, and we were able to control the precipitation and obtain fairly large poly-crystals.For longer spacers (dodecylamine to octadecylamine, C12–C18), we could not utilize the standard synthesis due to the lower solubility of these spacers in the acid. Our attempts included a significant decrease in the concentration, resulting in a very small amount of product and very small crystals.71 Heating up to even higher temperatures (under reflux) to compensate for the lower solubility and achieve the desired concentrations resulted in the deformation of the HI and the organic spacers. Even in cases where we managed to avoid the boiling of the solution and burning of the organic matter by slowing the rate of spacer addition, and thus allowing the synthesis to occur at slightly lower temperatures, the perovskite crystals that precipitated were too small to be filtered out of the solution. Taking the crystals directly out of the solution was also futile. Due to the large surface area of the 2D MHPs, the solution was not fully evaporated, and the outcome remained as a paste and not as poly-crystalline powder. As the excess solution affects the structural and optical characterization, we had to find a synthetic route that would allow us to overcome this issue.
To solve this issue, we utilized a different method – crystallization in the interphase between two solutions.72 In this method, Pb is dissolved in the HI solution, and a layer of methanol is added above, forming two separate liquid phases that are slowly mixed at the interface. The organic cations are dissolved in the minimal required volume of methanol, gently added to the pure methanol layer, and diffused through the interface, inducing the crystallization of MHPs. This procedure, occurring at room temperature, with no stirring, proved to be much more efficient with the longer ligands than the previous method used. This procedure was used to synthesize single crystals in previous works,72 so it was meant to be a slow precipitation. But with the long alkyl-ammonium ligands, the precipitation was a fairly fast process, with crystals starting to form in the interphase within minutes at most – or even immediately as it was added. Distinct and visible drops of the ligands’ solution “fell” through the interface as soon as it was added. This was associated with the different densities of the ligand solute compared to pure methanol. A possible solution is to decrease the concentration of the ligand, which can be achieved by decreasing the amount of ligands or increasing the volume of methanol. We chose the latter to maintain the quantity of 2D MHPs produced. Indeed, we found that significantly increasing the amount of methanol in which the ligands were dissolved led to slower diffusion of the ligands from the methanol solution, thus slowing the precipitation of the 2D MHPs. Since a smaller concentration of the ligands was added each time, they dissolved more evenly in the methanol solution on top of the HI. As a result of the better distribution of the ligands, the crystals formed slower, and larger crystals were obtained even for the longest ligands (Fig. 1A). We should note that the crystals synthesized using the longer spacers (C ≥ 10) formed thinner crystals in comparison to crystals synthesized using the shorter spacer, probably due to the low-symmetry arrangement of the longer spacers.
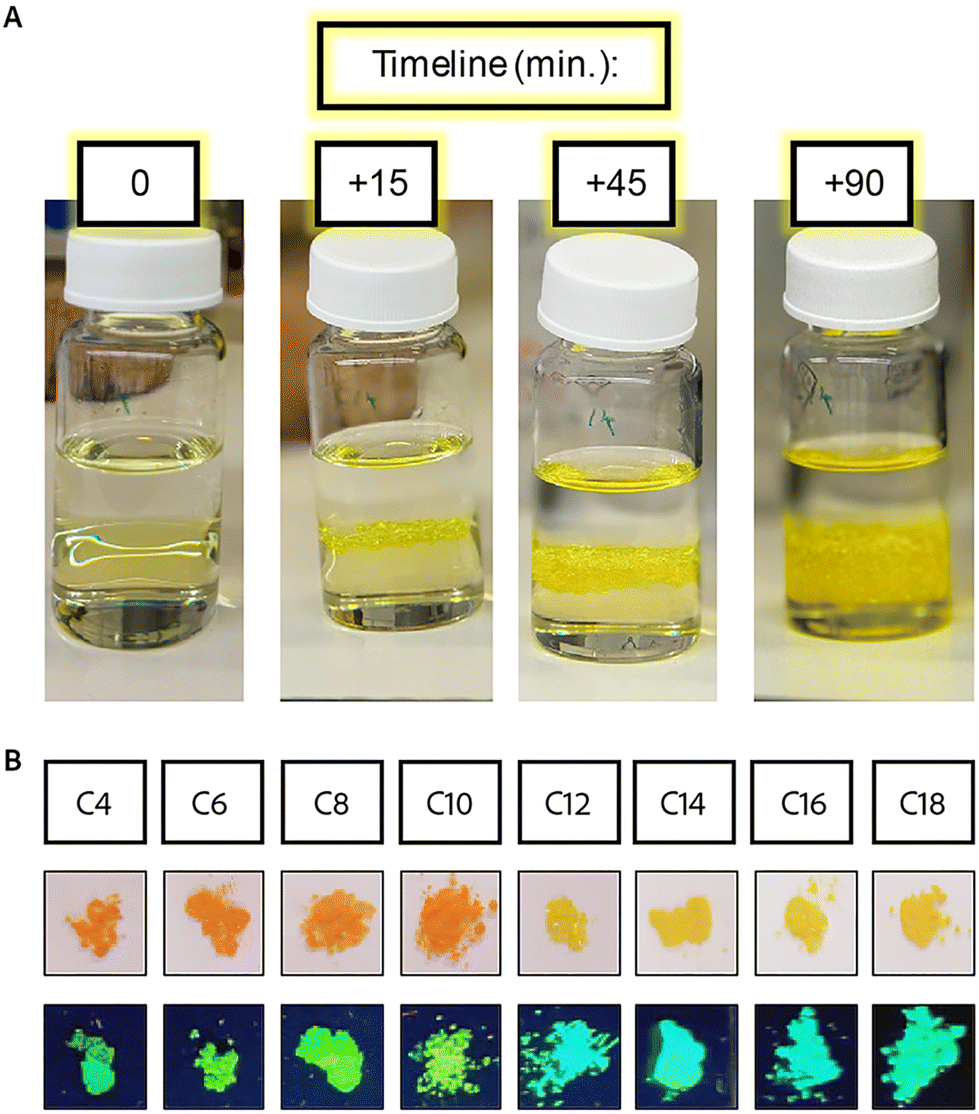 | ||
| Fig. 1 (A) Images taken during the synthesis of poly-crystals with long ligands (C ≥ 12) growing in the interphase between methanol and HI. (B) Images of the obtained poly-crystals under white light, and under UV light (bottom). | ||
Upon the successful integration of methanol, we tried to use other solvents that may enable better dissolution of the longer ligands (hexane, toluene, acetonitrile, diethyl ether). Although some materials precipitated in the interface with these solvents, the crystals were not as large as the methanol-grown ones, and we could not filter them from the solution. This highlights the importance of mixing between methanol and HI, forming an interphase volume that allows the growth of larger crystals.
All the 2D MHP crystals were formed as wide and thin plates, big enough to be filtered out from the solution using a standard sintered glass filter. The crystals formed with shorter ligands (C4, C6, and C8) were formed as thicker crystals (stacked ordered plates) than the rest of the series. The crystals of C4 to C10 are orange, while those of C12 to C18 are yellow (Fig. 1B). These colors fit the description of the previously reported structures characterized by Billing et al.,14,29,65 including the expected phase and color changes with temperature. Heating the yellow crystals caused them to turn orange, while cooling the crystals with C10 as barriers to ∼20 °C caused a change in the color to yellow. All of the crystals returned to their original color at room temperature. These phase transitions are associated with the transition towards higher symmetry and order of the spacers’ carbon chains with increased temperature. The higher order of the carbon affects the symmetry of the 2D inorganic layer, leading to higher order and specifically lower distortion of the octahedral corner sharing angles, turning the structures from monoclinic (at low temperatures) to orthorhombic (at elevated temperatures). The crystal structures were verified with pXRD measurements (Fig. S6†). For the samples with longer ligands (C12–C18), the peaks became broader, and some splitting was observed, associated with the phase transition to lower symmetry. The distance between the layers was calculated from the shifts in the angle of the first diffraction peak (Table S4†). The distance changes linearly as a function of the ligand length. However, due to the phase transition, we see two different slopes of the short ligands (C4–C10) and the long ligands (C12–C18), as highlighted in Fig. S6B.†
For the preparation of colloidal 2D NPs, we applied the ligand-assisted reprecipitation (LARP) method.56,58,68,70,73 Synthesizing colloids in this method requires dissolving bulk MHPs or stoichiometric ratios of precursor salts in DMF solution. Under vigorous stirring, a small amount of these solutions is swiftly injected into the antisolvent with or without additional ligands of the same type as in the structure, causing a fast reprecipitation. The fast precipitation leads to many nucleation grains and nano-sized crystals. We did not add any stabilizing ligands to the solution. After a short time, this solution was centrifuged, and the solution with the precursors was disposed to remove unreacted precursors from the solvent and prevent further crystallization. Then, the precipitated crystals were dispersed in a clean organic solvent and centrifuged again to separate larger crystals from the smaller ones. The supernatant was kept, and the precipitates were discarded. The main disadvantage of the LARP method is the difficulty of controlling the NP size and obtaining a narrow size distribution of the NPs. In our case, the NP sizes were distributed between 10's of nm to 1 μm (Fig. S7†).
During this study, we found that the choice of antisolvent depends on the nature of the spacers. We studied two different organic solvents – hexane and toluene. For C14–C18, the difference was insignificant. For C ≤ 10, using hexane led to the formation of a separate phase at the bottom of the vial, trapping the precursors inside the DMF, instead of mixing with hexane. We associated this phenomenon with the lower hydrophobicity of the shorter ligands, limiting their ability to act as efficient surfactants. The polarity difference between the solvent and the antisolvent played an important role in this case. Therefore, we decided to work with toluene as the antisolvent for all samples. Additional difference between the short and long ligands was the formation of significantly thicker NPs for the shorter ligands (C ≤ 10, Fig. S7†).
Temperature control was another important aspect of the colloidal synthesis with the shorter ligands. For the synthesis of colloids with C12 or shorter as a ligand, the room temperature variations significantly impacted the resultant crystal formation. For synthesis at temperatures of 16 °C–20 °C, all the colloidal NPs formed with the same color as the poly-crystals made with the same spacers (orange for C4–C10 and yellow for C12–C18). In contrast, in the cases of a higher ambient temperature (around 23 °C–25 °C), the colloidal products for C ≤ 12 formed in different structures from the poly-crystal, with a significant ‘red-shift’. For C12 with a typical yellow color, the NPs turned orange, and for C ≤ 10 with a typical orange color, the NPs turned red. These colors were obtained already within the first few seconds after the LARP injection. This red-shifted phase had much lower photoluminescence (PL). Cooling the synthesis with an ice bath helped stabilize the structures and prevent the color change. Freezing red-shifted samples for a short while in liquid nitrogen caused them to return temporarily to their original color and PL activity. These color changes correspond to the phase transitions described by Billing for poly-crystals.14,29,65 However, the observed phase transition temperatures for NPs were much lower when compared to those of the bulk compounds. Based on the bulk crystal characterization, the red phase had a smaller octahedral tilt, which led to better sharing of the atomic electronic wave functions, which was expected to result in improved mobility and a decrease in the PL intensity.74,75 Another outcome we observed with the ‘red-shifted’ NPs was that some of the samples became colorless within a few minutes after the injection. This process was irreversible and is associated with rapid material degradation; any optical activity (absorbance and PL) was lost.
Thin-film fabrication began with cleaning the substrates to obtain an even surface with minimal disturbance and repeatable wetting, which is crucial to obtain smooth films with large grains and a narrow size distribution. Briefly, substrates were washed and sonicated with soap water, dried with acetone and ethanol, and underwent ozone-plasma cleaning (see the Experimental section for full details). We used solutions similar to those used for the LARP synthesis described above for the film's deposition, obtained by dissolving pre-made 2D MHP polycrystals in DMF. These solutions were dispersed via spin-coating and annealed on a hot plate at 100 °C. Like with the polycrystals, the films of C12–C18 were orange when heated on the hot plate and turned back to the yellow phase when lifted and cooled back to room temperature. While thin films are generally bulkier than colloids, their preparation, much like colloids, is based on kinetic growth. Spin-coating a thin layer and curing on the hot plate lead to rapid precipitation of the crystals. This is different from the thermodynamic growth of poly-crystals and probably dictates the differences in the optical properties, as we observed.
Optical characterization
The initial characterization of the obtained 2D MHPs was through absorption spectroscopy, which could indicate the band gap energy and nature. The absorption is generally measured through the transmittance or reflection, and the ‘missing’ photons were calculated based on eqn (1).| I0 = IT + IR + IA. | (1) |
 | ||
| Fig. 2 Absorbance of the 2D (Cn)2PbI4 MHP. (A) Absorbance spectra of C4–C18, as poly-crystals, thin-films and colloids, and (B) comparison of the exciton wavelengths. | ||
For the non-transparent crystals (the polycrystals and the thin films), we used the IR, since in these cases IT ≪ IR. This is realized by measuring the diffusive reflectance of the samples using an integrating sphere. Information about the absorption was then obtained by applying the Kubelka–Munk (KM) conversion,76 given in eqn (2):
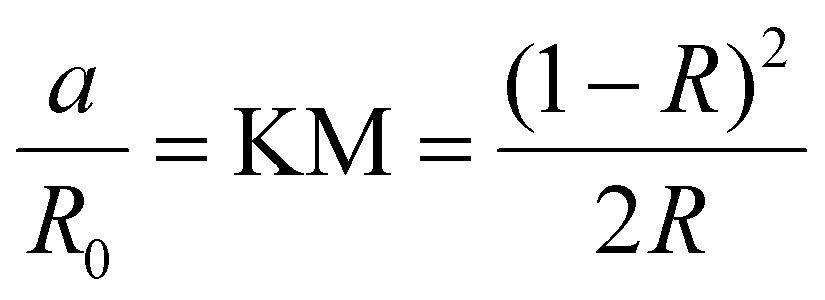 | (2) |
Analyzing the absorbance, we found that between the short (C ≤ 10) and the long spacers (C ≥ 12), there is a blue shift of ∼20 nm (Fig. 2A and B), matching with the observed color shift and the crystallographic changes.14,29,65 Based on the crystallographic analysis, the blue-shifted materials have stronger octahedral tilting on their shared plane. These octahedral angle distortions cause decreased charge carrier mobility and an increase in the energy of the band gap. When comparing the wavelengths of the excitons in each form of crystals (poly-crystals, thin films, and colloids), a red-shift was observed between the poly-crystals and the colloids of the same spacers, which decreases with the elongation of the ligands (Fig. 2B). This shift may result from the difference in the ratio of bulk (layers of crystals covered with other layers) vs. surface (exposed layers) in the different forms, which correlates with the results published by Nag et al.77 There was a visible difference in the thickness of the crystals, with C4–C8 being relatively thicker and C14–C18 seeming very thin plates. Since absorbance measures the lowest band gap in the sample, it cannot easily differentiate between the different components. This means that if there is a considerable presence of the bulk phase, it will probably cause a red shift in the absorbance.
Another aspect that arises when comparing the excitons of the three morphologies is that for shorter spacers (C4–C12), the thin films behave like the colloids, but as the ligands elongate (C ≥ 14), the films behave more like polycrystals. It may indicate that while the poly-crystalline structure tends to stack, the morphology of thin films prevents the stacking and the creation of ‘bulk areas’. Once the structure tends towards smaller and thinner particles (colloidal NPs), the formation of the films forces the creation of thicker layers (compared to the NPs), more similar to the poly-crystalline samples with the same spacers.
Steady-state PL
Similar to the absorbance measurements, the steady-state PL also showed a similar blue shift of the yellow structures (C12–C18) (Fig. 3A). The shift in PL energy was significant enough to see with the naked eye under UV light (Fig. 1B). This blue shift is an additional support to the increase of the band gap energy measured by absorbance, leading to higher energy of the emitted photons.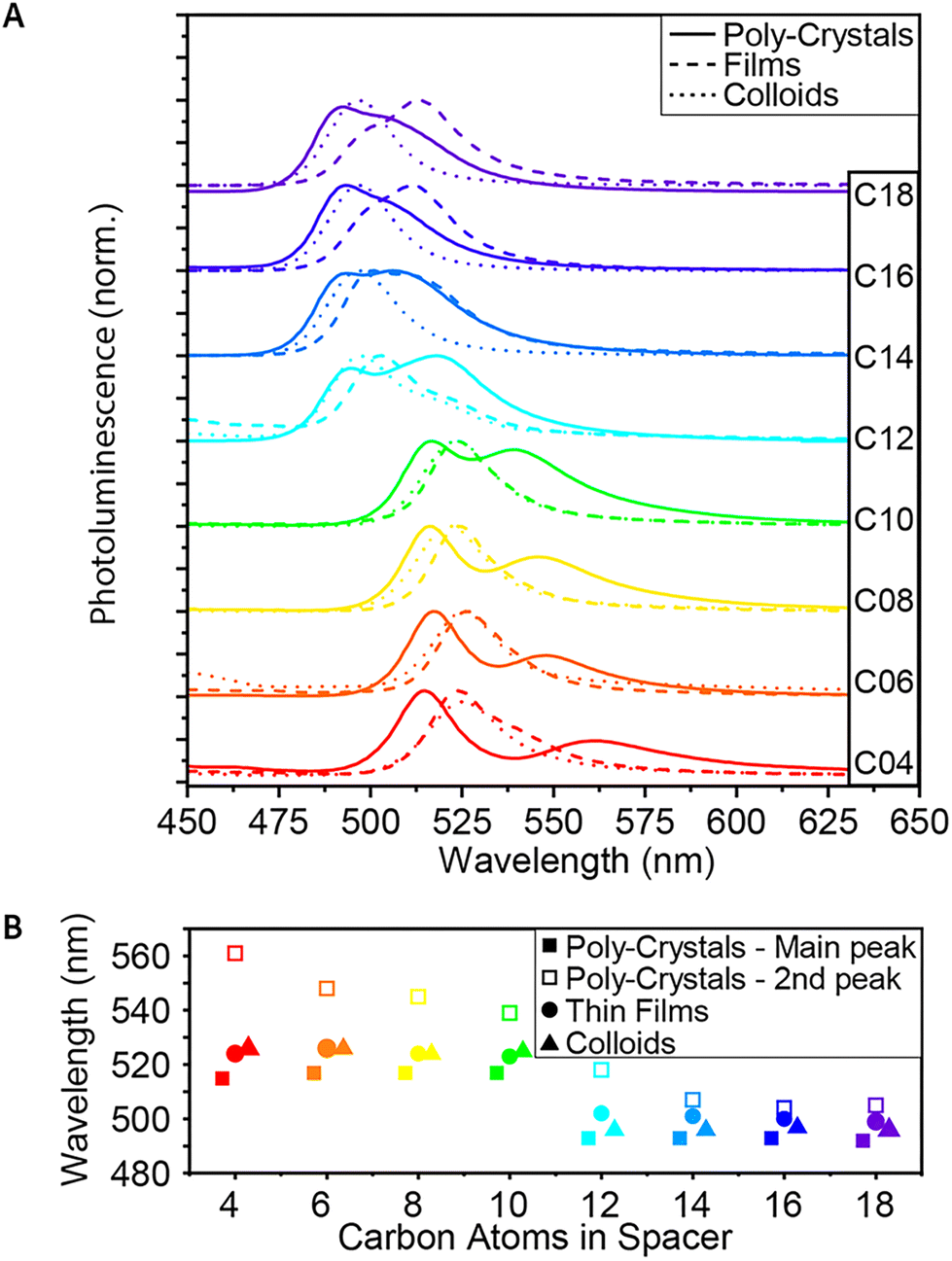 | ||
| Fig. 3 Steady-state PL of the 2D (Cn)2PbI4 MHP. (A) PL spectra of C4–C18, as poly-crystals, thin-films and colloids. (B) Comparison of the PL peak wavelengths. | ||
For the poly-crystalline samples, a secondary peak, lower in energy, was observed (Fig. 3A and B). This peak is ‘blue-shifted’ (higher in energy) with the elongation of the spacers and almost merged with the main peak in the case of the longest spacers. While the primary PL peak is more energetic than the lowest energy exciton of each of the poly-crystals, the secondary peak seems to be red-shifted relatively to the exciton peak (Fig. S1†). On the other hand, if we compare the main PL peak to a secondary peak observed in all of the absorbance spectra, we find a correlation between them, with an almost constant red-shift – 20–25 nm – from the second absorbance peak to the primary PL peak. Together with the blue shift of the absorbance peaks of the colloids relative to the peaks of the poly-crystals, this may imply that this more energetic absorbance peak is associated with the surface of the crystals.
In contrast to the poly-crystals, the colloids show only one PL peak. This indicates the more homogeneous structure of the small and thin colloids, mainly consisting of the ‘surface’ and much less of the ‘bulk’ phase. The energy of this PL peak is located in between the two peaks of the poly-crystals, as described above. For C4–C10, the shift is ∼10 nm, and for C12–C18, it is only shifted by ∼3 nm. This is another indication that for the longer spacers, the poly-crystals become more similar to the colloids.
While in the case of C4–C10, the PL curve of the thin films behaves the same as the PL curve of the colloids; starting with C12, we see a change (Fig. 3A and B). First, a secondary peak – like the one observed in the poly-crystals – appears and becomes much more significant as the spacer elongates. For C16 and C18, it is even more significant than the primary peak. In addition, unlike the PL peak of the colloids, which becomes more similar to the peak of the polycrystals, for the thin films, the shift remains ∼10 nm throughout the whole series. This correlates with our hypothesis, based on absorption measurements, that the film formation forces a more colloid-like structure for the shorter ligands and a more poly-crystal-like structure for the longer ones.
Time-resolved PL
Time-resolved PL (TRPL) was also measured in order to study the excited state dynamics and find the typical decay time of the PL. These measurements were taken using a micro-spectroscopy setup equipped with a pulsed laser, with a repetition rate of 10 MHz, enabling a measurement window of 100 ns, allowing sufficient time for the excited charges to decay. The emitted photons pass through an interferometer and are collected by using a fast single photon detector, enabling to resolve two-dimensional maps of the TRPL emission energy and decay time. It is important to indicate that unlike the steady-state PL and many ensemble PL measurements taken at a 45° angle, these measurements were taken at a 90° angle through an objective. This implies different and much stronger excitation compared to the steady-state PL.77 Hence, the observed TRPL spectra differ from those of the steady-state PL. Specifically, we could not identify the secondary peak in the TRPL spectra for the poly-crystals. This could be related to the measurement's angle or to the beam's higher intensity. Additional differences that may be related to these factors are a red-shift of ∼6 nm for the poly-crystals and a blue shift of ∼3 nm for the films. Another aspect we observed is that for the TRPL, there is a better correlation between the PL peaks of the poly-crystals and the films (Fig. S5†).Analyzing the 2D TRPL maps, we were able to observe different phases in the PL that change in time. To investigate this, we plot the PL spectra of three different temporal regions – short time (up to 2 ns after excitation), medium time (2–10 ns), and long time (10–60 ns). We observed that for the medium time, there was a mild red-shift (∼3 nm) in the PL peak of the poly-crystals (Fig. 4A, C and E). After this time frame, the peak returned to its original wavelength. Unlike poly-crystals, most of the colloidal samples did not show such a shift (Fig. 4B, D and F), which led us to conclude that this shift is related to the bulk area of the crystals. 2D MHP poly-crystalline structures have a significant bulk portion, especially compared to colloid crystals. While most of the PL in these measurements originates from the surface (mainly because of the angle), some photons may have penetrated the bulk. When most of the PL decayed, it is possible that delayed PL of the inner layers had some effects on the sum of the observed PL. We should note that the integrated PL intensity is magnitudes lower for the medium and long times compared to its peak intensity (at short times).
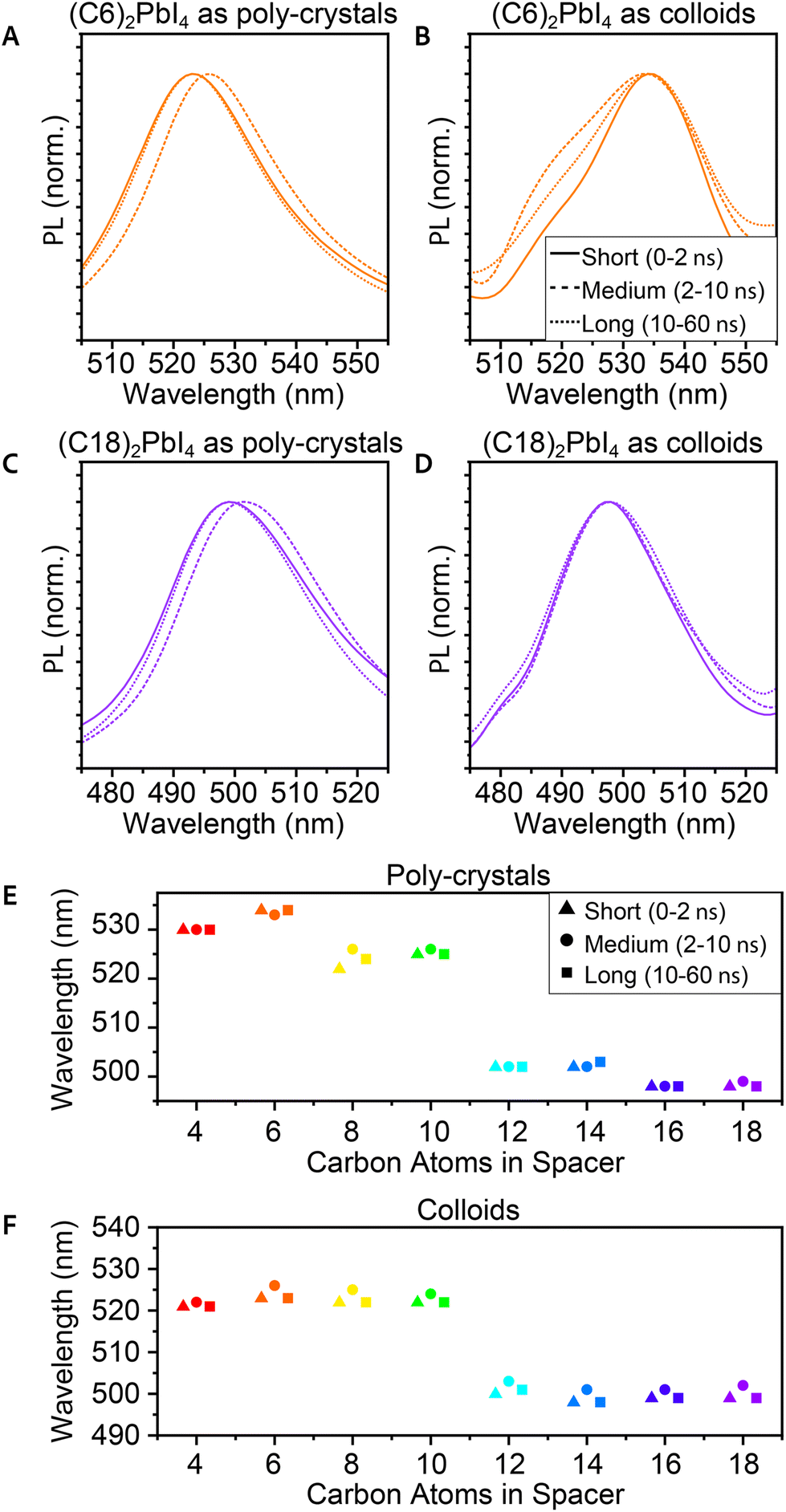 | ||
| Fig. 4 TRPL of the 2D (Cn)2PbI4 MHP in 3 time frames of the PL decay – 0–2 ns, 2–10 ns, and 10–60 ns. (A) (C6)2PbI4 as poly-crystals, and (B) as colloids. (C) (C18)2PbI4 as poly-crystals, and (D) as colloids. (E) Comparison of the PL peak wavelengths at the different time frames of poly-crystals and (F) colloids. | ||
One aspect in which we could not see a clear trend of change with the structural and morphology modifications was the lifetime of the PL decay (Fig. 5). The typical decay times were all below 1 ns, and we could not identify any clear trend. For all samples, the fitted lifetime gave an average decay constant faster than 1 ns, and for most samples, it varied between 400 ps and 700 ps; yet, as mentioned above, no clear trend was observed with the morphology of crystals or the spacer length. Since PL lifetime is dominated by defects in the structure, finding a correlation between the different aspects and the lifetime will require some optimizations of all the procedures. Although structural changes could imply a change in this property, finding a correlation to the emission lifetime will require optimization of the crystal preparation procedures.
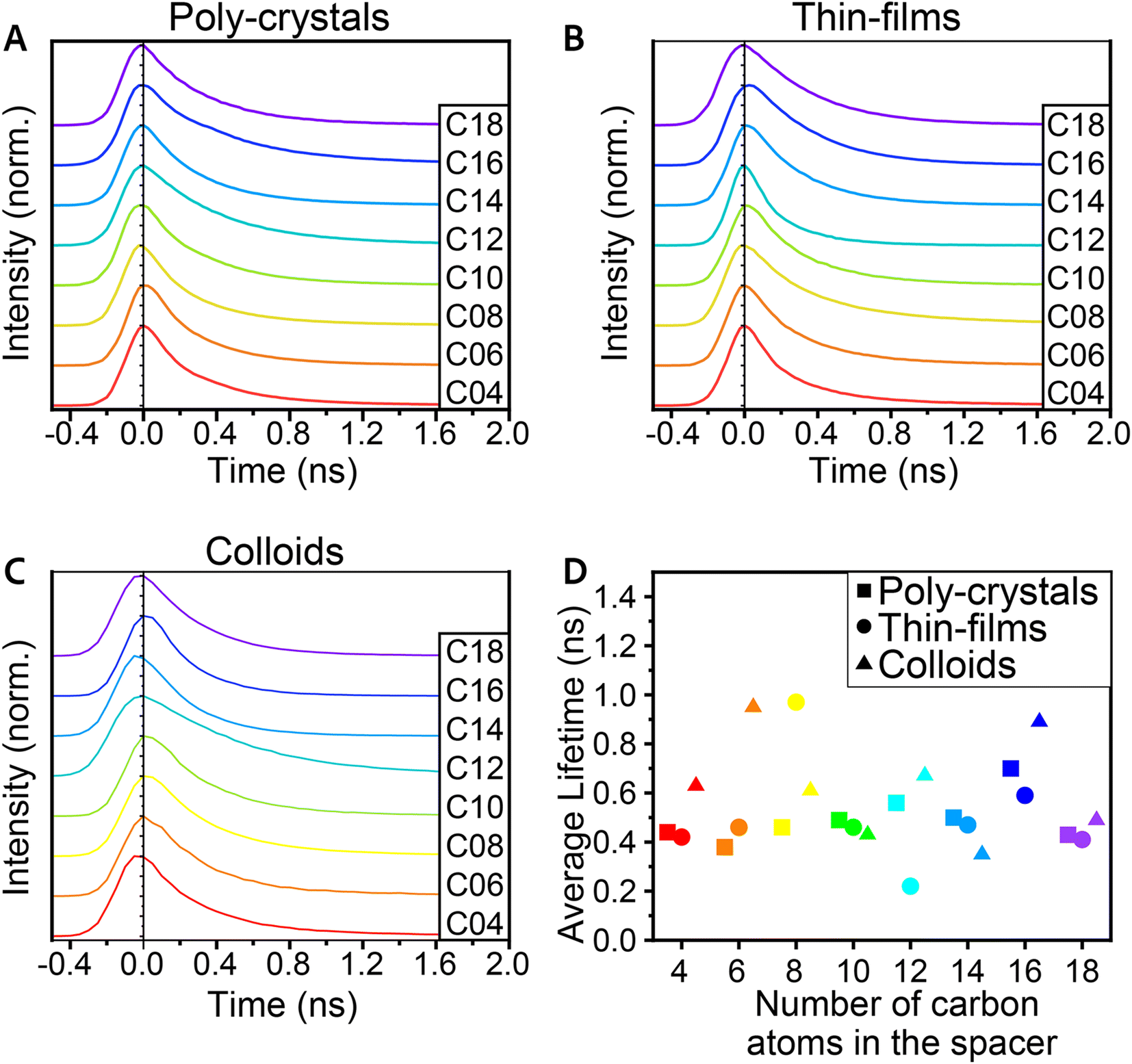 | ||
| Fig. 5 PL decay spectra of the 2D (Cn)2PbI4 MHP for (A) poly-crystals, (B) thin-films, and (C) colloids. (D) Averaged lifetime of PL decay for the poly-crystals, thin-films, and colloids. | ||
Summary
In this manuscript, we studied the structural and optical characteristics of 2D MHPs with different morphologies ranging from bulk poly-crystals to thin films and colloidal NPs. The observed optical and structural differences mainly relate to the crystals’ morphology and specifically thickness and bulk-to-surface ratio.In general, the synthesis of 2D MHPs is modified based on the spacer length and morphology. For bulk poly-crystals with longer spacers, an interphase synthesis combining polar and non-polar solvents was required due to the poor solubility of the longer spacers in HI. For colloidal synthesis, the phase transition temperatures were altered towards lower temperatures. For the shorter ligands, these phase transitions occur near room temperature, requiring cooling the reaction vessel during synthesis to ensure the formation of 2D colloids with the appropriate yellow/orange phase.
We noticed that 2D MHPs with longer spacers (C > 10) tend to form thinner crystals (plates-like) compared to the shorter ligand equivalents (C ≤ 10), for both poly-crystals and colloids. For thin films, we indicate that the morphology and thickness mainly depend on the fabrication process. Thin films fabricated using the described parameters resulted in thinner-than-expected structures for the short ligands (resembling colloids) and thicker-than-expected structures for the long ligands (more poly-crystal-like structures).
The optical properties of the 2D MHPs mainly depend on the structural distortion of the octahedral plane and the bulk-to-surface ratio. For all compounds, we observed a phase transition between the orange (orthorhombic) to yellow (monoclinic) structure upon cooling. As the spacers are elongated, this phase transition occurred at higher temperatures. For colloids, the phase transition temperatures were significantly lower compared to the bulk poly-crystals, and a less distorted red phase was observed, specifically for colloids with short ligands. The yellow phase is characterized by blue-shifted absorption and PL compared to the orange phase, of approximately 145 meV (490 nm to 520 nm).
A clear difference between bulk poly-crystals and colloids is the bulk-to-surface ratio. For poly-crystals, we see a lower energy absorption onset and two distinct PL peaks, which are associated with the bulk (lower energy) and surface (higher energy) portions of the 2D MHPs. For colloids and thin films, we observed a single PL peak and a blue shift of the absorption onset compared to the bulk poly-crystals (with equivalent spacers). For poly-crystals, as the spacers were elongated, a thinner morphology (lower bulk-to-surface ratio) was observed, and the optical properties became similar to the colloids.
Experimental
Synthesis
Characterization
Author contributions
Conceptualization: O. A. and I. H.; investigation: O. A., R. H. and L. A.; validation: E. M. and I. H.; visualization: O. A., E. M. and I. H.; supervision: I. H.; writing: O. A., E. M. and I. H.Data availability
The data supporting this article have been included as part of the ESI.† Any additional data are available through Ido Hadar (HUJI).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Zelman Cowen Academic Initiatives (ZCAI-HUJI joint projects).References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- I. Chung, B. Lee, J. He, R. P. H. Chang and M. G. Kanatzidis, Nature, 2012, 485, 486–489 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- Y. Chen, Y. Sun, J. Peng, J. Tang, K. Zheng and Z. Liang, Adv. Mater., 2018, 30, 1703487 CrossRef PubMed.
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS PubMed.
- C. Li, H. Sun, S. Gan, D. Dou and L. Li, Mater. Futures, 2023, 2, 042101 CrossRef CAS.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52(15), 9019–9038 CrossRef CAS PubMed.
- Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties | Inorganic Chemistry, https://pubs.acs.org/doi/full/10.1021/ic401215x, (accessed 29 December 2024).
- P. Basumatary, J. Kumari and P. Agarwal, Optik, 2022, 266, 169586 CrossRef CAS.
- W. Zhang, G. E. Eperon and H. J. Snaith, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121, 2230–2291 CrossRef CAS PubMed.
- D. G. Billing and A. Lemmerer, New J. Chem., 2008, 32, 1736–1746 RSC.
- P. Vashishtha, M. Ng, S. B. Shivarudraiah and J. E. Halpert, Chem. Mater., 2019, 31, 83–89 CrossRef CAS.
- M. Rahil, R. M. Ansari, C. Prakash, S. S. Islam, A. Dixit and S. Ahmad, Sci. Rep., 2022, 12, 2176 CrossRef CAS.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- K. Wang, C. Wu, D. Yang, Y. Jiang and S. Priya, ACS Nano, 2018, 12, 4919–4929 CrossRef CAS PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS.
- Z. S. Almutawah, S. C. Watthage, Z. Song, R. H. Ahangharnejhad, K. K. Subedi, N. Shrestha, A. B. Phillips, Y. Yan, R. J. Ellingson and M. J. Heben, MRS Adv., 2018, 3, 3237–3242 CrossRef CAS.
- N. Kitazawa, M. Aono and Y. Watanabe, J. Phys. Chem. Solids, 2011, 72, 1467–1471 CrossRef CAS.
- Y. Zhang and Y. Zhou, ACS Energy Lett., 2024, 9, 186–190 CrossRef CAS.
- M. Lu, Y. Zhang, S. Wang, J. Guo, W. W. Yu and A. L. Rogach, Adv. Funct. Mater., 2019, 29, 1902008 CrossRef.
- D. B. Mitzi, Chem. Mater., 1996, 8, 791–800 CrossRef CAS.
- S. E. Chang, C.-Y. Park, E. Yoon, J. S. Kim and T.-W. Lee, J. Inf. Disp., 2024, 25, 97–120 CrossRef CAS.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, C. M. M. Soe, J. Yoo, J. Crochet, S. Tretiak, J. Even, A. Sadhanala, G. Azzellino, R. Brenes, P. M. Ajayan, V. Bulović, S. D. Stranks, R. H. Friend, M. G. Kanatzidis and A. D. Mohite, Adv. Mater., 2018, 30, 1704217 CrossRef.
- C. C. Stoumpos, D. H. Cao, D. J. Clark, J. Young, J. M. Rondinelli, J. I. Jang, J. T. Hupp and M. G. Kanatzidis, Chem. Mater., 2016, 28, 2852–2867 CrossRef CAS.
- D. G. Billing and A. Lemmerer, Acta Crystallogr., Sect. B:Struct. Sci., 2007, 63, 735–747 CrossRef CAS PubMed.
- N. Kitazawa, M. Aono and Y. Watanabe, J. Phys. Chem. Solids, 2011, 72, 1467–1471 CrossRef CAS.
- H. Nagasaka, M. Yoshizawa-Fujita, Y. Takeoka and M. Rikukawa, ACS Omega, 2018, 3, 18925–18929 CrossRef CAS PubMed.
- I. Spanopoulos, I. Hadar, W. Ke, Q. Tu, M. Chen, H. Tsai, Y. He, G. Shekhawat, V. P. Dravid, M. R. Wasielewski, A. D. Mohite, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 5518–5534 CrossRef CAS PubMed.
- L. Zhu, D. Liu, J. Wang and N. Wang, J. Phys. Chem. Lett., 2020, 11, 8502–8510 CrossRef CAS PubMed.
- X. Chen, H. Zhou and H. Wang, Front. Chem., 2021, 9 DOI:10.3389/fchem.2021.715157.
- S. Zeng, S. Shi, S. Wang and Y. Xiao, J. Mater. Chem. C, 2020, 8, 1319–1325 RSC.
- T. Liu, Z. Zhang, Q. Wei, B. Wang, K. Wang, J. Guo, C. Liang, D. Zhao, S. Chen, Y. Tang, Y. Zhou and G. Xing, Chem. Eng. J., 2021, 411, 128511 CrossRef CAS.
- Y. Li, Y. Lei, H. Wang and Z. Jin, Nano-Micro Lett., 2023, 15, 128 CrossRef CAS PubMed.
- M. Han, Y. Xiao, C. Zhou, Y. Yang, X. Wu, Q. Hu, X. Jin, W. Zhang, J.-S. Hu and Y. Jiang, Mater. Futures, 2023, 2, 012104 CrossRef CAS.
- V. B. Mykhaylyk, H. Kraus and M. Saliba, Mater. Horiz., 2019, 6, 1740–1747 RSC.
- P. S. Mathew, J. T. DuBose, J. Cho and P. V. Kamat, ACS Energy Lett., 2021, 6, 2499–2501 CrossRef CAS.
- T. Zhu, Y. Yang, K. Gu, C. Liu, J. Zheng and X. Gong, ACS Appl. Mater. Interfaces, 2020, 12, 51744–51755 CrossRef CAS PubMed.
- Q. Tu, D. Kim, M. Shyikh and M. G. Kanatzidis, Matter, 2021, 4, 2765–2809 CrossRef CAS.
- S. He, L. Qiu, L. K. Ono and Y. Qi, Mater. Sci. Eng., R, 2020, 140, 100545 CrossRef.
- E.-B. Kim, M. S. Akhtar, H.-S. Shin, S. Ameen and M. K. Nazeeruddin, J. Photochem. Photobiol., C, 2021, 48, 100405 CrossRef CAS.
- J. Shi, Y. Gao, X. Gao, Y. Zhang, J. Zhang, X. Jing and M. Shao, Adv. Mater., 2019, 31, 1901673 CrossRef PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- Q. A. Akkerman, T. P. T. Nguyen, S. C. Boehme, F. Montanarella, D. N. Dirin, P. Wechsler, F. Beiglböck, G. Rainò, R. Erni, C. Katan, J. Even and M. V. Kovalenko, Science, 2022, 377, 1406–1412 CrossRef CAS PubMed.
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS PubMed.
- E. Scharf, F. Krieg, O. Elimelech, M. Oded, A. Levi, D. N. Dirin, M. V. Kovalenko and U. Banin, Nano Lett., 2022, 22, 4340–4346 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- J. Butkus, P. Vashishtha, K. Chen, J. K. Gallaher, S. K. K. Prasad, D. Z. Metin, G. Laufersky, N. Gaston, J. E. Halpert and J. M. Hodgkiss, Chem. Mater., 2017, 29, 3644–3652 CrossRef CAS.
- A. Swarnkar, R. Chulliyil, V. K. Ravi, M. Irfanullah, A. Chowdhury and A. Nag, Angew. Chem., 2015, 127, 15644–15648 CrossRef.
- F. Zhang, H. Zhong, C. Chen, X. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- C. J. Dahlman, N. R. Venkatesan, P. T. Corona, R. M. Kennard, L. Mao, N. C. Smith, J. Zhang, R. Seshadri, M. E. Helgeson and M. L. Chabinyc, ACS Nano, 2020, 14, 11294–11308 CrossRef CAS PubMed.
- J. A. Sichert, Y. Tong, N. Mutz, M. Vollmer, S. Fischer, K. Z. Milowska, R. García Cortadella, B. Nickel, C. Cardenas-Daw, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6521–6527 CrossRef CAS PubMed.
- Q. A. Akkerman, S. G. Motti, A. R. Srimath Kandada, E. Mosconi, V. D'Innocenzo, G. Bertoni, S. Marras, B. A. Kamino, L. Miranda, F. De Angelis, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 1010–1016 CrossRef CAS PubMed.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS PubMed.
- R. Chakraborty and A. Nag, J. Phys. Chem. C, 2020, 124, 16177–16185 CrossRef CAS.
- A. N. Yadav, S. Min, H. Choe, J. Park and J. Cho, Small, 2024, 20, 2305546 CrossRef CAS PubMed.
- E. H. Massasa, R. Strassberg, R. Shechter, S. Dror, S. Khalfin, S. Shaek, M. K. Khristosov, I. Hadar and Y. Bekenstein, Adv. Funct. Mater., 2024, 34, 2311122 CrossRef CAS.
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS PubMed.
- A. Lemmerer and D. G. Billing, Dalton Trans., 2012, 41, 1146–1157 RSC.
- K. Vighnesh, S. Wang, H. Liu and A. L. Rogach, ACS Nano, 2022, 16, 19618–19625 CrossRef CAS PubMed.
- R. A. Dziatko, S. M. Chintapalli, Y. Song, E. Daskopoulou, D. E. Kachman, Z. Zander, D. L. Kuhn, S. M. Thon and A. E. Bragg, J. Phys. Chem. Lett., 2024, 15, 4117–4124 CrossRef CAS PubMed.
- J.-H. Wei, X.-D. Wang, J.-F. Liao and D.-B. Kuang, ACS Appl. Electron. Mater., 2020, 2, 2707–2715 CrossRef CAS.
- L. C. Schmidt, A. Pertegás, S. González-Carrero, O. Malinkiewicz, S. Agouram, G. Mínguez Espallargas, H. J. Bolink, R. E. Galian and J. Pérez-Prieto, J. Am. Chem. Soc., 2014, 136, 850–853 CrossRef CAS PubMed.
- H. Huang, Y. Li, Y. Tong, E.-P. Yao, M. W. Feil, A. F. Richter, M. Döblinger, A. L. Rogach, J. Feldmann and L. Polavarapu, Angew. Chem., Int. Ed., 2019, 58, 16558–16562 CrossRef CAS PubMed.
- T. Sheikh, V. Nawale, N. Pathoor, C. Phadnis, A. Chowdhury and A. Nag, Angew. Chem., Int. Ed., 2020, 59, 11653–11659 CrossRef CAS PubMed.
- M.-H. Tremblay, J. Bacsa, B. Zhao, F. Pulvirenti, S. Barlow and S. R. Marder, Chem. Mater., 2019, 31, 6145–6153 CrossRef CAS.
- V. A. Hintermayr, A. F. Richter, F. Ehrat, M. Döblinger, W. Vanderlinden, J. A. Sichert, Y. Tong, L. Polavarapu, J. Feldmann and A. S. Urban, Adv. Mater., 2016, 28, 9478–9485 CrossRef CAS PubMed.
- T. Ishihara, J. Takahashi and T. Goto, Phys. Rev. B:Condens. Matter Mater. Phys., 1990, 42, 11099–11107 CrossRef CAS PubMed.
- N. Kitazawa, M. Aono and Y. Watanabe, Thin Solid Films, 2010, 518, 3199–3203 CrossRef CAS.
- A. K. Roy Choudhury, in Principles of Colour and Appearance Measurement, ed. A. K. Roy Choudhury, Woodhead Publishing, Oxford, 2015, pp. 117–173 Search PubMed.
- T. Sheikh, A. Shinde, S. Mahamuni and A. Nag, ACS Energy Lett., 2018, 3, 2940–2946 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05516k |
| This journal is © The Royal Society of Chemistry 2025 |