
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic conversion of biomass-derived oxygenated aromatics to cycloalkanes†
Meheryar R.
Kasad
 ab,
James E.
Jackson
c and
Christopher M.
Saffron
*ab
ab,
James E.
Jackson
c and
Christopher M.
Saffron
*ab
aDepartment of Chemical Engineering and Materials Science, Michigan State University, East Lansing, Michigan 48824, USA
bDepartment of Biosystems and Agricultural Engineering, Michigan State University, East Lansing, Michigan 48824, USA. E-mail: saffronc@msu.edu
cDepartment of Chemistry, Michigan State University, East Lansing, Michigan 48824, USA
First published on 27th November 2024
Abstract
Electrocatalytic hydrotreatment (ECH) was explored as a mild technique to convert oxygenated aromatics, present in oils derived from the deconstruction of lignocellulosic biomass or lignin, into cycloalkanes. Producing cycloalkanes in a one-pot system, as envisioned in the present study, requires that both hydrodeoxygenation and aromatic ring saturation occur electrocatalytically. Thus, an activated carbon cloth-supported ruthenium and platinum (RuPt/ACC) electrocatalyst was synthesized and used to conduct model compound ECH studies to determine substrate conversion, product yields, and faradaic efficiency, enabling the derisking of the electrocatalytic process. The effects of electrocatalyst composition and aromatic ring substituents on cycloalkane yield were examined. Furthermore, ECH of side products and probable intermediates was conducted to map reaction sequences and pathways. Finally, ECH of a 4-O-5 dimer model compound was conducted to study the electrocatalytic cleavage of recalcitrant interunit linkages in lignin.
Introduction
The adverse effects of greenhouse gas emissions from the combustion of petroleum products for transportation,1,2 coupled with energy security and independence considerations,3 have encouraged a shift towards the use of fuels (and chemicals) derived from renewable lignocellulosic biomass in recent years. Lignin constitutes up to 30 wt% of lignocellulosic biomass and is a renewable source of aromatic compounds.4 Lignin can be isolated from lignocellulosic biomass and deconstructed for the production of fuels and chemicals. Deconstruction of lignin by fast pyrolysis yields a lignin pyrolysis oil, which is closely related to the “bio-oil” produced by fast pyrolysis of lignocellulosic biomass. The volatile fraction of lignin pyrolysis oil (and bio-oil) consists of monoaromatic phenolic compounds, including phenols, guaiacols, and syringols. These volatile phenolics, detectable by gas chromatography mass spectrometry (GC-MS), constitute up to 3.8 wt% of bio-oils.5 Reductive catalytic fractionation (RCF) of lignocellulosic biomass has recently emerged as a promising technique for valorizing the lignin fraction of biomass. This process yields lignin oil that is primarily composed of a narrow set of phenolic monomers and functionally similar dimers and oligomers.6,7Complete hydrodeoxygenation (HDO) and aromatic ring saturation of the lignin-derived oxygenated aromatic (phenolic) compounds result in the formation of cycloalkanes. These “green” cycloalkanes could potentially be blended into jet fuels (C8–C16). Nominally, monocycloalkanes constitute 20–25 wt% of Jet A8,9 and Jet A-1 (ref. 8) fuels derived from kerosene. Importantly, Muldoon and Harvey identified bio-based cycloalkanes as the missing link for the production of full-performance and ultraperformance drop-in synthetic jet fuels.10 The density, freeze point, flash point, and specific energy of monocycloalkanes can exceed conventional fuel requirements.11 One strategy for producing cycloalkanes from phenolic compounds is reductive upgrading by electrocatalytic pathways. Electrocatalytic hydrotreatment (ECH) offers several advantages over conventional thermocatalytic pathways. First, the required hydrogen equivalents are produced in situ by water splitting, avoiding the use of flammable hydrogen gas (H2), which poses safety concerns, particularly at small-scale regional depots. Furthermore, in the present market, H2 is primarily derived from fossil resources such as natural gas and coal.12 Secondly, this energy upgrading process can be powered by renewable electricity generated from solar and wind energy. Thirdly, the chemical transformations are achieved under relatively mild conditions (temperatures ≤ 60 °C and atmospheric pressure), compared to the high temperatures and hydrogen pressures required for similar transformations in conventional thermocatalytic processes. Thus, the proposed electrocatalytic processes could potentially be implemented at regional depots near biomass production areas.
The electrocatalytic conversion of lignin-derived phenolic compounds to cycloalkanes has been reported in a few studies. Zhao et al. obtained cyclohexane upon ECH of phenol on graphite-supported platinum (Pt/graphite) in acidic catholyte solutions (H2SO4, HCl and HClO4).13 Meanwhile, Liu et al. used a dual-catalyst electrochemical system comprising a suspended noble-metal catalyst (i.e., Pt/C, Pd/C, Rh/C, and Pt/Al2O3) and a soluble polyoxometalate (e.g., silicotungstic acid) for HDO of bio-oil model compounds.14 Phenolic compound HDO was achieved at high current densities and high faradaic efficiencies using this setup. High selectivity towards cycloalkanes was achieved, particularly on Pt/C, with 28% methylcyclohexane selectivity from 4-methylphenol reduction. Wijaya et al. employed a stirred slurry catalytic reactor for the electrocatalytic HDO of phenolic compounds.15 However, the three-component electrolyte system (methanesulfonic acid, propan-2-ol, and NaCl or KCl) used in this study did not include an electron transfer catalyst. Nonetheless, high cycloalkane selectivities (up to 40%) and high faradaic efficiencies were obtained in this setup. Interestingly, Zhai et al. employed a fluidized electrochemical system consisting of phosphotungstic acid, suspended noble-metal catalyst (i.e. Pt/C, Ru/C, Rh/C, and Pd/C) and sodium borohydride (NaBH4).16 This setup involved both reagent-based and electrocatalytic reduction, resulting in the formation of cycloalkane products from phenolic compounds. High cycloalkane selectivities and high faradaic efficiencies were obtained on Pt/C, with 60% propylcyclohexane selectivity and 92% faradaic efficiency achieved during 2-methoxy-4-propylphenol reduction. More recently, Han et al. used the same approach to conduct a comparative analysis on the ECH of G and S-type phenolic model compounds.17 Relatively lower cycloalkane yields from ECH of phenolic compounds have also been reported on ruthenium supported on activated carbon cloth (Ru/ACC)18 and various platinum-based catalysts (Pt, PtNi, PtNiB, and Pt/NiB) supported on ordered mesoporous carbon (CMK-3).19
Overall, most of the reported studies on conversion of phenolic compounds to cycloalkanes by ECH were conducted in stirred slurry (fluidized) reactors, which offer several advantages over conventional H-cells where the electrocatalysts are present in the form of wires, foils, or metals dispersed on conductive supports. However, electron transfer in stirred slurry reactors requires the use of expensive electrolytes (electron transfer agents) or direct contact between the suspended particles and the electrode surface, conditions that can be challenging to achieve when processing concentrated solutions of viscous oils at a commercial scale. Therefore, the present study seeks to investigate the conversion of oxygenated aromatic compounds to cycloalkanes in a conventional H-cell setup, where the noble metal catalyst particles are dispersed on a carbon-based support that also serves as the electrode.
In the present study, a bimetallic electrocatalyst, RuPt/ACC, comprising ruthenium and platinum supported on activated carbon cloth, was developed to investigate the ECH of several lignin-derived oxygenated aromatic compounds. The relative loading of the two metals on the activated carbon cloth support was varied to identify the composition that maximized propylcyclohexane yield from 4-propylguaiacol (4-PrGu) ECH. To determine the effect of substituents on the product slate, ECH of 4-alkyl/allylguaiacols and 4-allylsyringol was also conducted. Furthermore, ECH of the likely intermediates and side products was undertaken to map reaction sequences and pathways. Finally, the conversion of di-p-tolyl ether, a model dimer, to methylcyclohexane by ECH was investigated.
Experimental
Electrocatalyst preparation
The electrocatalyst was prepared by incipient wetness impregnation followed by reduction with H2. Ruthenium precursor solution (0.22 M Ru basis) was prepared by dissolving hexaammineruthenium(III) chloride [Ru(NH3)6Cl3] in aqua ammonia [28.0–30.0% NH3 basis] (19.6% volume fraction) and deionized water. Chloroplatinic acid (8 wt% in water) served as the platinum precursor solution. Activated carbon cloth [ACC] (Zorflex®∼Double Weave Activated Carbon Fiber Cloth, Charcoal House, Crawford, NE) pieces cut to 1.5 cm × 3.0 cm were immersed in deionized water overnight and then oven dried at 150 °C for 1 hour. Requisite amounts of the two precursor solutions were then sequentially added to the washed and dried ACC pieces with intermittent drying at ambient conditions. The ACC pieces impregnated with Ru and Pt precursors were dried overnight at ambient conditions, then vacuum dried for 24 h. These vacuum-dried ACC pieces, impregnated with the Ru and Pt precursors, were then reduced under H2 in a 300 ml Parr Mini Bench Top Reactor (Parr Instrument Company, Moline, IL) at 300 °C and approximately 650 psi. The reduced RuPt/ACC pieces were stored in a desiccator prior to use in the ECH trials.Electrocatalyst characterization
The surface area and pore volume of the electrocatalysts were determined by nitrogen (N2) physisorption at 77 K (Micromeritics® 2020 ASAP). The samples were outgassed for 24 h at 130 °C prior to the analysis.Scanning electron microscopy (SEM) was used to determine the morphology of the electrocatalyst. The images were collected on a JSM-6610 LV (JEOL Ltd) scanning electron microscope. Energy dispersive X-ray spectroscopy (elemental analysis) was performed using an Oxford Instruments AZtec system (Oxford Instruments, High Wycomb, Bucks, England), software version 3.3 using a 20 mm2 Silicon Drift Detector (JSM 6610LV) and an ultra-thin window.
The Ru and Pt contents of the Ru/ACC, Pt/ACC and RuPt/ACC electrocatalysts were determined by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES). The analyses were conducted on an Agilent 5800 ICP-OES instrument equipped with AVS 6/7 and SPS 4 Autosampler. The samples were digested in a mixture of concentrated nitric acid and hydrochloric acid (volume ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) using the CEM Mars 6 Microwave Digestion System. Calibration solutions were prepared from ruthenium(III) chloride (RuCl3) and chloroplatinic acid standard solutions (Inorganic Ventures, Christiansburg, VA).
:1) using the CEM Mars 6 Microwave Digestion System. Calibration solutions were prepared from ruthenium(III) chloride (RuCl3) and chloroplatinic acid standard solutions (Inorganic Ventures, Christiansburg, VA).
ECH setup
ECH experiments were conducted in batch mode in a glass H-cell fabricated by the Chemistry Glass Shop at Michigan State University (East Lansing, MI). The two chambers were separated using a Nafion™ 117 (Chemours, Wilmington, DE) membrane. RuPt/ACC was the cathode, while the anode was a platinum wire (99.95%; Strem Chemicals Newburyport, MA). A schematic representation of the ECH setup is shown in Fig. 1. A sulfuric acid (H2SO4) solution (0.67 M) with 10% (volume fraction) propan-2-ol was used as the electrolyte solution in both compartments. The H-cell assembly was immersed in a heated water bath to maintain the desired temperature (50/60 °C). The ECH trials for oxygenated aromatics with methyl substituents were conducted at 50 °C to minimize the loss of methylcyclohexane, the desired product, due to evaporation and gas stripping. The RuPt/ACC cathode was subjected to a pre-ECH reduction step at 80 mA for 10 min. After this, the requisite amount of a substrate stock solution was added to the cathode compartment, resulting in a catholyte solution with a substrate concentration of 0.02 M (or 0.01 M for dimers) at the start of a trial. Both compartments contained 13 ml of electrolyte solution. A Xantrex XHR 300–3.5 DC power supply supplied constant electric current throughout the trial. | ||
| Fig. 1 ECH of 4-alkylguaiacol in a two-chambered H-cell. The reactions and flow of electrons and protons are depicted. Rubber stoppers with needles were placed on top of the anode and cathode compartments to allow continuous venting of the gases produced at the electrodes. The solutions in both compartments were agitated using magnetic stir bars. | ||
Sample analysis
Catholyte solution samples collected at the end of each trial were saturated with sodium chloride (NaCl) and extracted into dichloromethane (DCM). 1.00 ml of catholyte solution was saturated with 400 mg NaCl and extracted with 2.0 ml DCM. The cathode (RuPt/ACC) was also extracted in DCM (5.0 ml) and the resulting DCM solution was dried using sodium sulfate (Na2SO4). The DCM extracts were analyzed using GC-MS (Shimadzu QP 5050A). Standard solutions of the reactants and products in DCM were used to construct calibration curves for quantifying sample concentrations. Cumene was added as an internal standard for the quantification of cycloalkanes, while octan-2-ol was added as an internal standard for quantification of oxygenated compounds. The sample concentrations thus determined were used to calculate reactant conversion, product yields and faradaic efficiency.List of compound/functional group abbreviations
A list of abbreviations for compounds and functional groups relevant to the present study is provided in Table 1.| Compound/functional group | Abbreviation |
|---|---|
| Phenol | PhOH |
| Anisole [methoxybenzene] | PhOMe |
| Guaiacol [IUPAC: 2-methoxyphenol] | Gu |
| Syringol [IUPAC: 2,6-dimethoxyphenol] | Sy |
| Cyclohexane | ChH |
| Cyclohexan-1-ol [cyclohexanol] | ChOH |
| Cyclohexan-1-one [cyclohexanone] | Ch![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
| Di-p-tolyl ether | Tol2O |
| Alkyl | R |
| Methyl | Me |
| Ethyl | Et |
| Propyl | Pr |
| Allyl | Ay |
| Methoxy | MeO |
Calculations
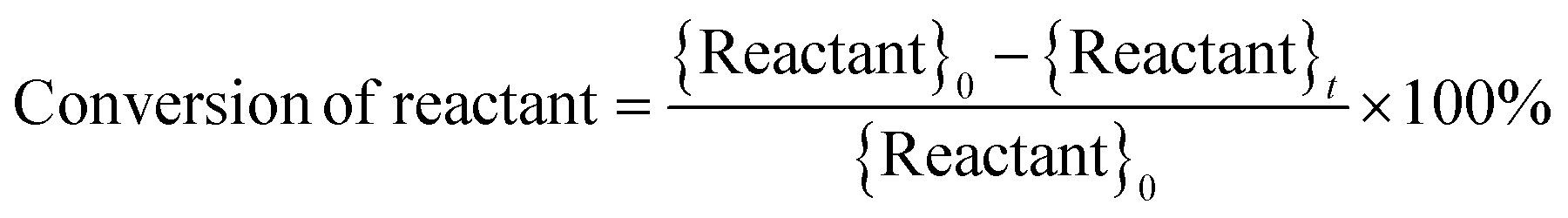
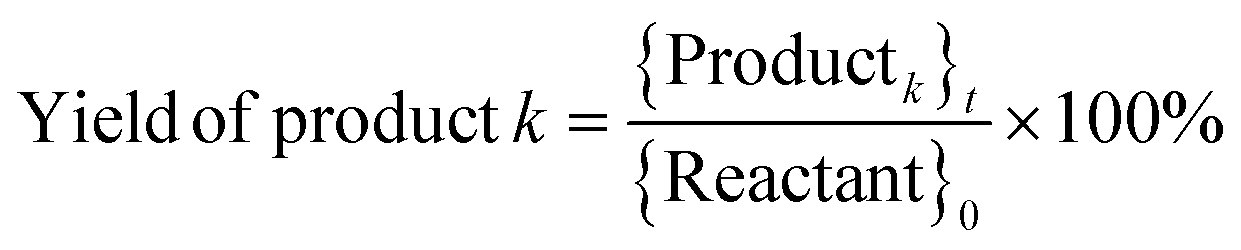
where, {Reactant}0 is the amount of reactant at the start of a trial (time, t = 0) (in mol). {Reactant}t is the amount of reactant that remains unreacted at the end of a trial (in mol). {Productk}t is the amount of product k formed at the end of a trial (in mol). Faraday's constant, F = 96
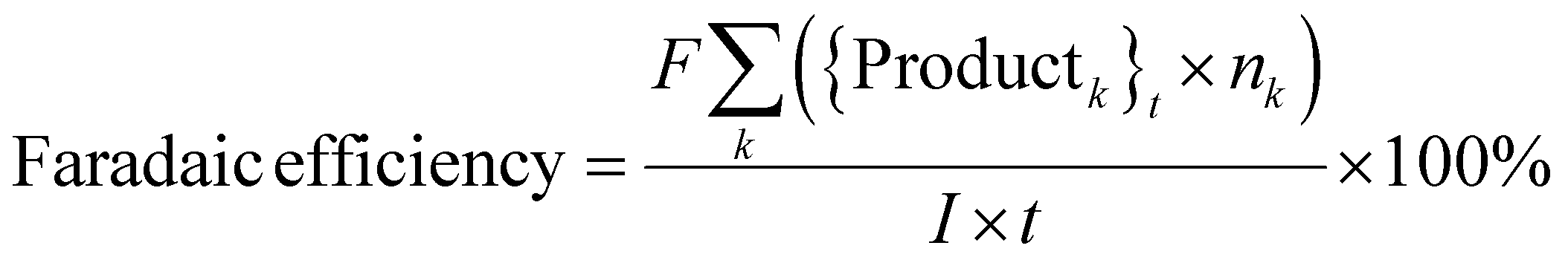 485.3 C mol−1. nk is the number of electrons (e−) required to form product k. I is the current. t is the trial run-time.
485.3 C mol−1. nk is the number of electrons (e−) required to form product k. I is the current. t is the trial run-time.
Note that the faradaic efficiencies were computed with respect to the yield of recovered products. The reported faradaic efficiencies do not account for H2 formation, products that may have formed but were lost due to evaporation and gas stripping, and products formed in trace quantities whose yields were not quantified.
Results and discussion
Electrocatalyst characterization
The washed and dried ACC, Ru/ACC, Pt/ACC and RuPt/ACC samples were characterized by N2 physisorption at 77 K. BET surface area (SBET) was calculated in the relative pressure range, p/p0 < 0.06 (where, p is the equilibrium pressure and p0 is the saturation vapor pressure), determined using the criteria developed by Rouquerol et al.20 for microporous adsorbents. The Gurvich rule was used to estimate the total pore volume (Vt) based on the amount of vapor adsorbed at a relative pressure close to unity.3 The Dubinin–Radushkevich equation was used to determine the micropore volume (Vμpore) in the following relative pressure range: 10−4 < p/p0 < 0.02.22 The average pore width (Dp) was calculated using the equation: Dp = 4Vt/SBET. The results are summarized in Table S1 of the ESI.† The blocking of some pores of the ACC by Ru and Pt could explain the relatively lower BET surface area, total pore volume, and micropore volume of Ru/ACC, RuPt/ACC and Pt/ACC compared to the washed and dried ACC.SEM images of the Ru0.75Pt0.25/ACC electrocatalyst are shown in Fig. 2. It is observed in Fig. 2(a) that the electrocatalyst synthesis procedure results in dispersal of the noble metal (Ru and Pt) particles on the activated carbon cloth fibers. However, the SEM images collected at higher magnification (Fig. 2(b) and (c)) reveal a non-uniform morphology of the noble metal particles. Broadly, the noble metal particles are present as standalone cube-like structures or clusters of needle-like structures. The elemental map acquired by EDS (see Fig. S1†) suggests close association between Ru and Pt and does not indicate their dispersal at separate sites.
 | ||
| Fig. 2 SEM images of Ru0.75Pt0.25/ACC. (a) Magnification: 35× reveals dispersal of the noble metal particles and (b) and (c) magnification: 600× reveals morphology of noble metal particles. | ||
The Ru/ACC, RuPt/ACC, and Pt/ACC samples were digested in a solution composed of 3 parts concentrated nitric acid (HNO3) and 1 part concentrated hydrochloric acid (HCl) at 230 °C for analysis by ICP-OES. A solid residue was obtained upon digestion of Ru-containing samples, suggesting incomplete digestion in the acid solution. To confirm that the solid residue was not derived from the ACC support, washed and dried ACC samples were subjected to digestion in the acid solution at 230 °C. Additionally, no solid residue was obtained upon digestion of the Pt/ACC samples, indicating that the residual solids contained Ru. Moreover, the resistance of Ru (particularly, Ru metal and anhydrous RuO2) to chemical attack by acids has been reported in the literature.23,24 Significantly, no solid residue was obtained upon digestion of vacuum-dried Ru/ACC, Pt/ACC, and RuPt/ACC samples that had not been reduced under H2, in the acid solution at 230 °C. Therefore, the solutions obtained upon digestion of the vacuum-dried (unreduced) samples were also analyzed by ICP-OES. The loadings for the vacuum-dried samples do not account for potential losses during reduction under H2; therefore, the values represent an upper bound for the expected loading. The results are summarized in Table 2.
| Ru content (wt. basis) | Pt content (wt. basis) | Ru:Pt (mole basis)a |
|
|---|---|---|---|
| a The values enclosed in brackets represent the targeted Ru to Pt mole ratio for bimetallic electrocatalysts. b Denotes that the samples were not subjected to reduction under H2. | |||
| Ru1.0Pt0.0/ACC | 4.0% | — | — |
| Ru0.75Pt0.25/ACC | 3.7% | 3.3% | 2.1 [3.0] |
| Ru0.50Pt0.50/ACC | 2.8% | 5.9% | 0.9 [1.0] |
| Ru0.25Pt0.75/ACC | 1.7% | 8.8% | 0.4 [0.3] |
| Ru0.0Pt1.0/ACC | — | 13.0% | — |
| Ru1.0Pt0.0/ACCb | 6.6% | — | — |
| Ru0.75Pt0.25/ACCb | 5.0% | 2.8% | 3.5 [3.0] |
| Ru0.5Pt0.5/ACCb | 3.1% | 5.3% | 1.1 [1.0] |
| Ru0.25Pt0.75/ACCb | 1.6% | 8.0% | 0.4 [0.3] |
| Ru0.0Pt1.0/ACCb | — | 10.8% | — |
The Ru and Pt loadings reported above indicate that the Ru to Pt mole ratios for the vacuum-dried bimetallic electrocatalysts align with the mole ratios targeted during electrocatalyst synthesis. The Pt loading of the vacuum-dried samples is lower than that of the samples reduced under H2. This may be attributed to the presence of chloride ligands and hydronium counter ions from chloroplatinic acid, as well as ammonia ligands and chloride counter ions from Ru(NH3)6Cl3 in the vacuum-dried samples, which can evolve as HCl, NH3, and water vapor during reduction under H2, decreasing the overall weight of the reduced samples. Meanwhile, the Ru loading of the vacuum-dried samples is found to be lower than that of the samples reduced under H2 (except for Ru0.50Pt0.50/ACC and Ru0.25Pt0.75/ACC, where they are similar), which is attributed to the loss of Ru as a solid residue upon digestion of the reduced samples. Overall, the Ru and Pt loading determined by ICP-OES suggests that the electrocatalyst synthesis broadly results in an equivalent metallic loading (on a molar basis) on the ACC support, with variations in the relative amounts of the two metals. The determination of accurate Ru and Pt loadings in future investigations would require the development of alternative approaches such as fusion methods.
Effects of electrocatalyst composition (relative loading of Ru and Pt)
ECH of 4-propylguaicol (4-PrGu) [IUPAC: 2-methoxy-4-propylphenol] was conducted on Ru/ACC and Pt/ACC to compare the performance of these two noble metal-based electrocatalysts. Notwithstanding the higher oxophilicity of Ru,25,26 the HDO performance of Pt/ACC was superior to Ru/ACC in terms of both 4-PrGu conversion and the yields of propylcyclohexane (PrChH) and 4-propylcyclohexanol (4-PrChOH). To explore the potential enhancements in HDO and aromatic ring saturation during ECH, RuPt/ACC electrocatalysts were synthesized. The superior HDO capability of carbon-supported bimetallic catalysts for phenolic substrates has been reported in several thermocatalytic studies.27–30 Recall that the electrocatalysts have an equivalent metal loading (on a molar basis) on the ACC support, with different Ru to Pt ratios. The products obtained upon ECH were PrChH, 4-PrChOH and 2-methoxy-4-propylcyclohexan-1-ol (2-MeO-4-PrChOH), along with trace quantities of 4-propylcyclohexan-1-one (4-PrChO). The mass spectra of the unassigned peaks on the chromatogram indicated formation of 1-methoxy-3-propylcyclohexane (1-MeO-3-PrChH). However, its identity could not be verified due to non-availability of reference mass spectra and neat standards; thus, its yield was not quantified. The ECH reaction products are shown in Scheme 1 and the quantitation results for the five electrocatalysts are summarized in Fig. 3. The bimetallic electrocatalysts showed synergistic effects, outperforming both single metal electrocatalysts. This finding is consistent with a recent study by Page et al. that investigated electrocatalytic HDO of phenol on carbon-supported Ru and Pt electrocatalysts.31 Interestingly, the highest 4-PrGu conversion and PrChH yield were obtained on Ru0.75Pt0.25/ACC, the bimetallic combination with the lowest proportion of Pt. In a study on toluene hydrogenation to methylcylohexane, Fukazawa et al. proposed that the high activity of the RuPt electrocatalysts could be explained by the spillover of adsorbed H radicals from Pt to toluene, which was strongly adsorbed on Ru.32 A similar mechanism was also proposed for hydrogenation of 4-substituted benzoic acids on RuPt electrocatalysts.33 On the other hand, Du et al. indicated based on in situ Raman spectroscopy and theoretical calculations that modification of the electronic structure of Pt by Ru was responsible for the synergistic effect observed during hydrogenation of benzoic acid derivatives on RuPt electrocatalysts.34 Thus, determination of the underlying mechanism for the synergies seen in the present study will be important in future work, including consideration of added complexity due to the deoxygenation reactions.
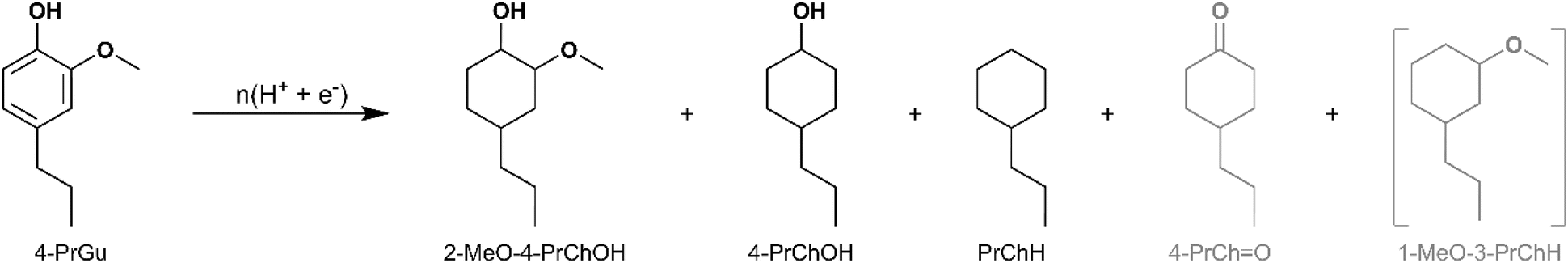 | ||
| Scheme 1 Products observed for ECH of 4-PrGu. Yield quantification was not performed for compounds in grey. The formation of compounds enclosed in brackets is inferred but their identity was not confirmed. Two peaks representing the isomers (cis and trans) of 4-PrChOH were observed by GC-MS. Three peaks representing the isomers of 2-MeO-4-PrChOH were observed by GC-MS, but quantitation was performed only for the two peaks observed by GC-MS for the external standard. | ||
 | ||
| Fig. 3 ECH of 4-PrGu. 4-PrGu conversion, product yields and faradaic efficiency. Experimental conditions: current = 60 mA; (mol e−)/(mol 4-PrGu) = 25; temperature = 60 °C. Error bars represent standard errors. | ||
Effects of alkyl/allyl substituents at the para position of guaiacol
The influence of methyl, ethyl, and allyl substituents at the para position (relative to the hydroxy group) of guaiacol on the ECH product slate was investigated using Ru0.75Pt0.25/ACC. The results were compared with 4-PrGu ECH. Both 4-methylguaiacol (4-MeGu) and 4-ethylguaiacol (4-EtGu) were converted by ECH to the analogous alkylcyclohexane (RChH) and 4-alkylcyclohexan-1-ol (4-RChOH), along with trace quantities of 4-alkylcyclohexan-1-one (4-RChO). The mass spectra of the unassigned peaks on the chromatogram indicated formation of 3-alkyl-1-methoxycyclohexane and 4-alkyl-2-methoxycyclohexan-1-ol. However, their identity could not be verified due to the non-availability of reference mass spectra and neat standards; thus, the yields were not quantified. The ECH reaction products are shown in Scheme 2 and the quantitation results are summarized in Fig. 4. The RChH yield from 4-EtGu was less than that from 4-PrGu, while the 4-RChOH yield from 4-EtGu was slightly higher than that from 4-PrGu. Note that the ECH trials for both substrates were conducted at 60 °C. The RChH and 4-RChOH yields from 4-MeGu were similar to those from 4-EtGu, even though the 4-MeGu ECH trials were conducted at 50 °C. Eugenol [IUPAC: 2-methoxy-4-prop-2-enylphenol] ECH resulted in a similar product slate as 4-PrGu ECH. Notably, 4-PrGu was obtained as a product from eugenol ECH. The complete conversion of eugenol and the absence of the allyl functionality in the products indicate that saturation of the olefinic group on the allyl chain occurs in the initial stages of the batch reaction process. In fact, the DFT study by Huš et al. demonstrated that hydrogenation of the allyl group is the first step during eugenol HDO on Ru(0001).35 Notably, it was shown that this reaction occurs homogeneously in the absence of a catalyst. The significantly lower yield of PrChH from eugenol ECH compared to 4-PrGu ECH, notwithstanding the passage of proportionately higher charge through the circuit ((mol e−)/(mol substrate) was 25 for 4-PrGu and 30 for eugenol), suggests suppression of HDO by the olefinic group on the allyl substituent.
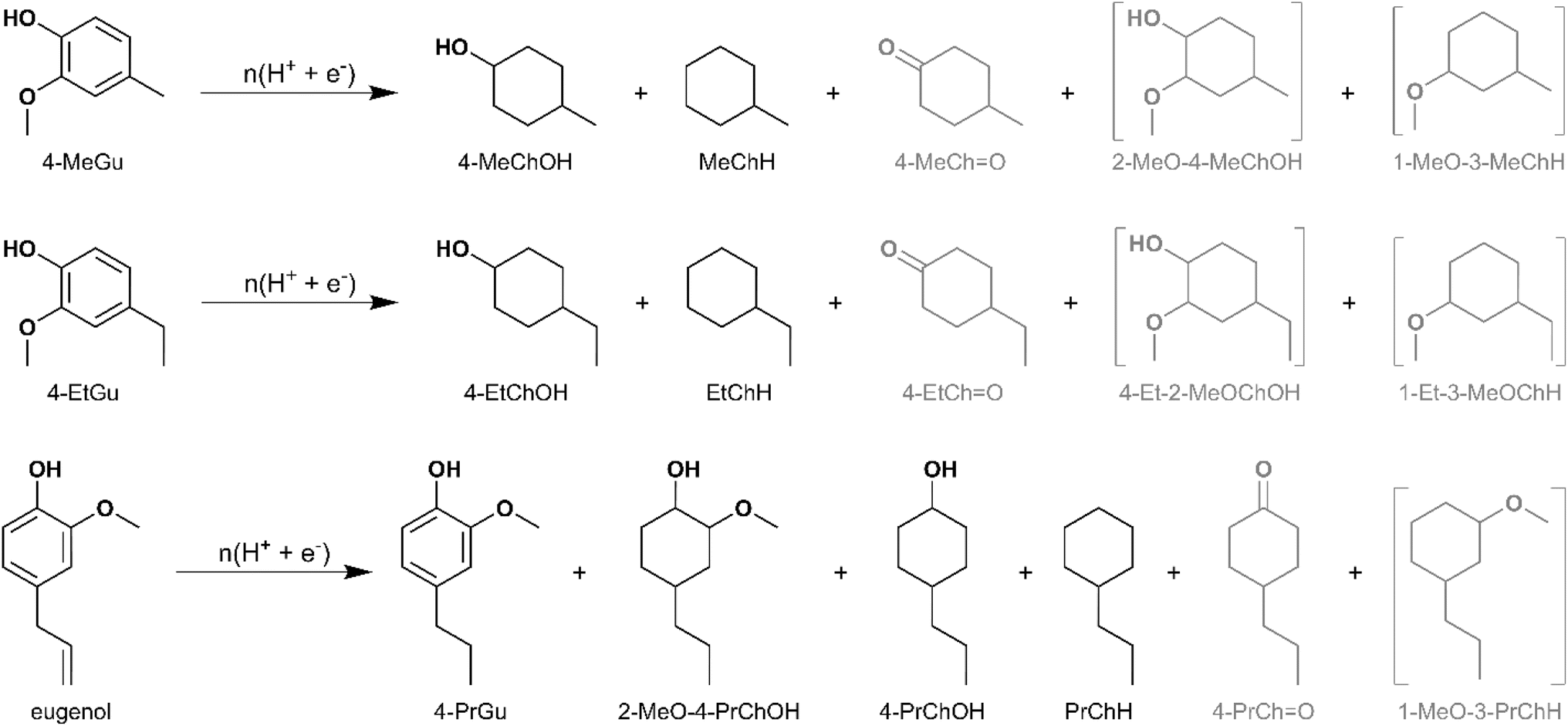 | ||
| Scheme 2 Products observed for ECH of 4-MeGu, 4-EtGu and eugenol. Yield quantification was not performed for compounds in grey. The formation of compounds enclosed in brackets is inferred but their identity was not confirmed. Two peaks representing the isomers (cis and trans) of 4-MeChOH, 4-EtChOH and 4-PrChOH were observed by GC-MS. Three peaks representing the isomers of 2-MeO-4-PrChOH were observed by GC-MS, but quantitation was performed only for the two peaks observed by GC-MS for the external standard. | ||
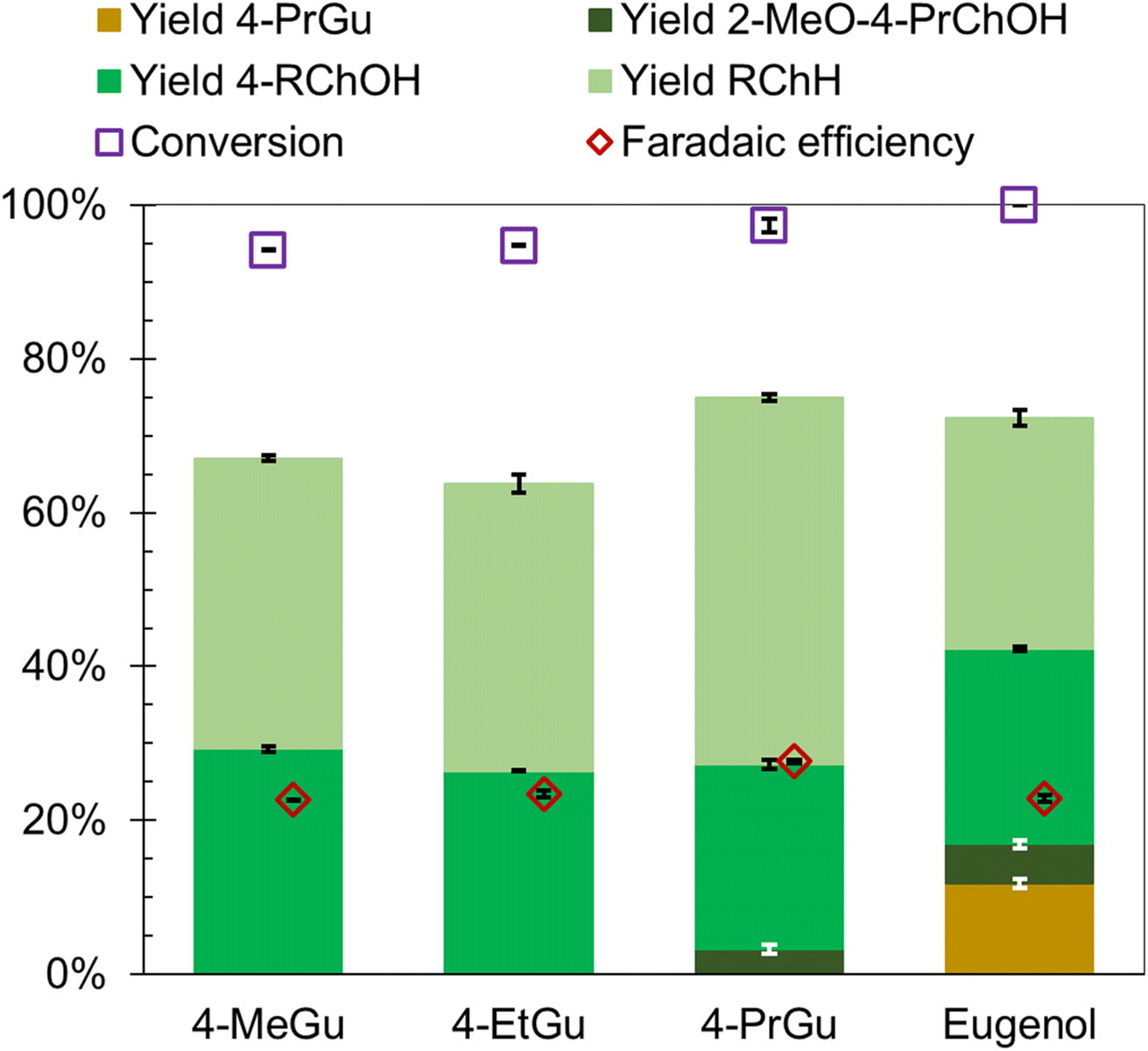 | ||
| Fig. 4 ECH of 4-MeGu, 4-EtGu, 4-PrGu and eugenol. Substrate conversion, product yields and faradaic efficiency. Experimental conditions: current = 60 mA; (mol e−)/(mol substrate) = 25 (4-MeGu, 4-EtGu and 4-PrGu) and 30 (eugenol); temperature = 50 °C (4-MeGu) and 60 °C (4-EtGu, 4-PrGu and eugenol). Error bars represent standard errors. | ||
Attempts to achieve mole balance closure and the results of the control experiment in the absence of noble metal loading on ACC are discussed in the mole balance closure section of the ESI.†
4-Alkylguaiacol ECH: reaction sequence and pathway mapping
It has been proposed that HDO of phenols and cresols (methylphenols) may occur via three distinct pathways, based on the sequence of individual steps (i.e., hydrogenation, tautomerization, C–O hydrogenolysis, and dehydration). These pathways are: direct deoxygenation (DDO), hydrogenation-deoxygenation (HYD) and tautomerization-hydrogenation-deoxygenation (TAU).36–41 The dominant pathway under a set of conditions is determined by the influence of various factors, including temperature, hydrogen pressure, surface affinity of different tautomers, catalyst (oxophilicity), support (acidity), and solvent (for liquid phase reactions).42 Typically, severe conditions are required for Caryl–O scission (DDO) due to the high bond dissociation energy (BDE). Saturation of the aromatic ring lowers the C–O BDE (see Table S2†), allowing dehydration reactions to occur on acidic catalyst supports (HYD).43–45 Accordingly, the TAU pathway (or an alternative involving partial aromatic ring hydrogenation) has been proposed as the dominant mechanism on less acidic catalyst supports.36,404-RGu contains three C–O bonds, C1–OaH, C2–ObC7 and C7–ObC2 as shown in Scheme 3, while alkylphenols and cresols contain a single C–O bond. 4-RGu conversion to RChH requires scission of two C–O bonds (i.e., C1–Oa and C2–Ob) and saturation of the aromatic ring. Consequently, ECH of the likely intermediates and side products of 4-RGu ECH was conducted to determine the reaction sequence and pathway for the formation of RChH from 4-RGu.
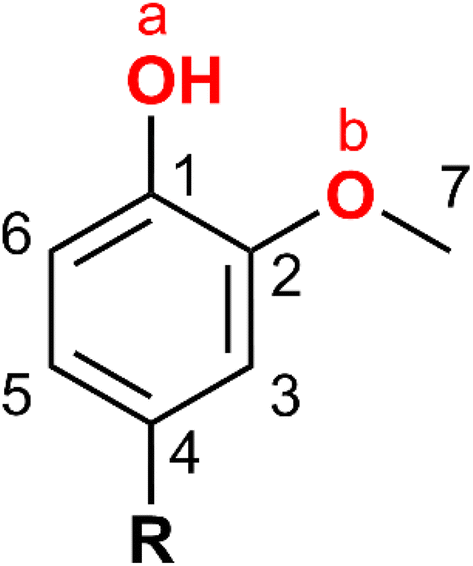 | ||
| Scheme 3 C–O bonds in 4-alkylguaiacol (4-RGu). | ||
Cyclohexanol (ChOH) formation in moderate-to-high yields from guaiacol (Gu) [IUPAC: 2-methoxyphenol] by ECH has been previously reported.18,19,46–49 Meanwhile, methoxycyclohexane formation has been reported in only one study,14 and even in that study, cyclohexanol selectivity was typically higher. These results suggest that methoxy group scission occurs more readily than hydroxy group scission during ECH. This also follows from the BDEs (see Table S2†) where the difference in energy required for cleavage of C–OH and C–OCH3 is around 10 kcal mol−1 for structurally similar compounds. Further evidence is provided in the study by Shangguan et al. which established that Gu HDO at the Ru nanoparticle–water interface proceeded via methoxy group cleavage based on rate assessments, H/D isotopic labeling, NMR spectroscopy, and first-principles calculations.50 The proposed mechanism involved H-adatom attack on the meta carbon of the sorbed guaiacol forming a partially saturated enol that underwent a kinetically relevant C–O bond cleavage step involving the shuttling of the hydroxyl proton to the methoxy leaving group by water, with concomitant stabilization of the transition state by the Ru center.
Along the DDO and TAU pathways, 4-alkylphenol and 3-alkylanisole were identified as likely intermediates depending on the sequence of Caryl–O bond cleavage. Therefore, ECH of 4-propylphenol (4-PrPhOH) and 4-propylanisole (4-PrPhOMe) was investigated in the present study. 4-PrPhOMe was selected as a model instead of 3-propylanisole since it could be readily procured. 4-PrPhOH was converted to PrChH and 4-PrChOH, along with trace quantities of 4-PrChO and propylbenzene, by ECH. Meanwhile 4-PrPhOMe primarily yielded PrChH, along with 4-PrChOH as a minor product, and trace quantities of 4-PrChO and propylbenzene. The mass spectra of the unassigned peaks on the chromatogram indicated formation of 4-propyl-1-methoxycyclohexane (4-Pr-1-MeOChH). However, its identity could not be verified due to non-availability of reference mass spectra or neat standards; thus, its yield was not quantified. The ECH reaction products are shown in Scheme 4 and the quantitation results are summarized in Fig. 5. The higher PrChH yield from 4-PrPhOMe compared to 4-PrPhOH indicates that demethoxylation was accomplished more readily compared to dehydroxylation. The conversion of 4-PrPhOMe to 4-PrChOH confirms the occurrence of a demethylation reaction step, which may involve electrocatalytic hydrogenolysis, hydrolysis, or C–H activation of the methyl group followed by hydrolysis (see Scheme 5). Notably, 4-alkylcatechol and 4-alkylcyclohexane-1,2-diol were not detected as products of 4-RGu ECH.
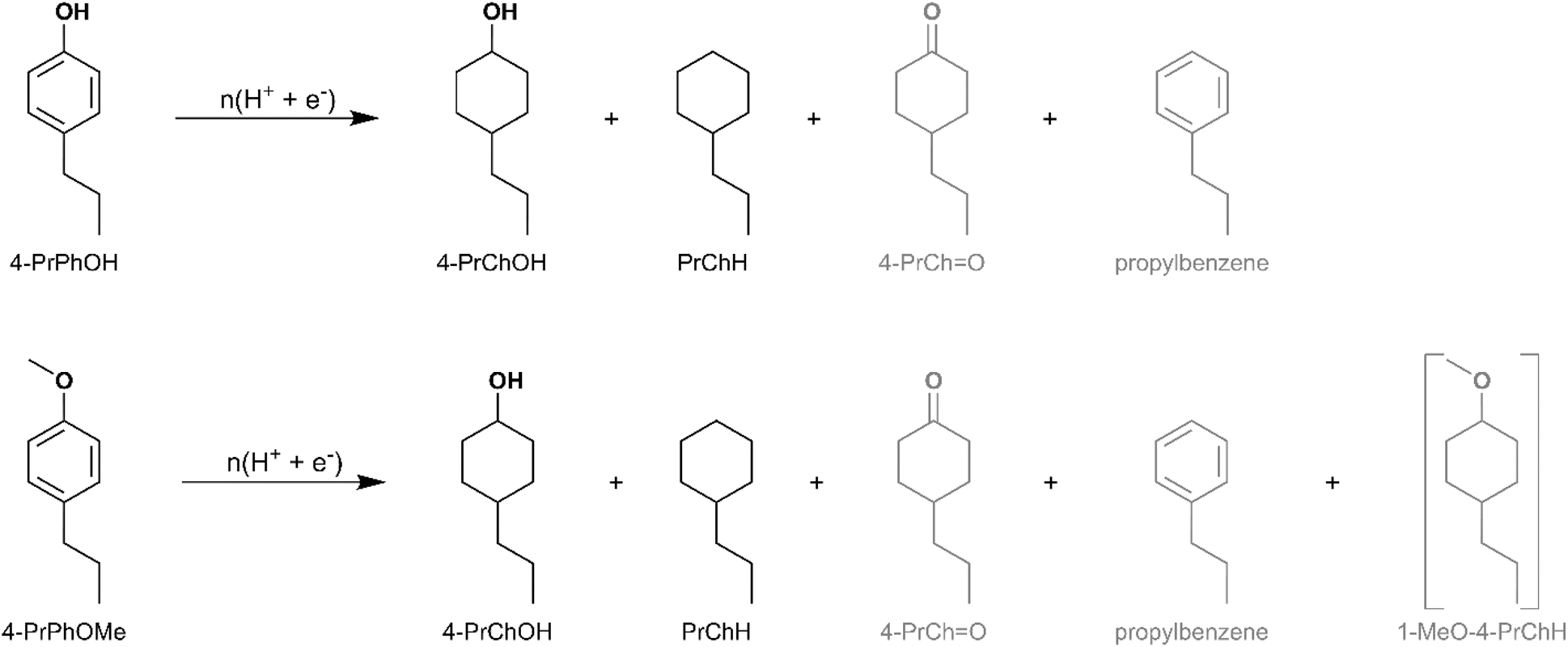 | ||
| Scheme 4 Products observed for ECH of 4-PrPhOH and 4-PrPhOMe. Yield quantification was not performed for compounds in grey. The formation of compounds enclosed in brackets is inferred but their identity was not confirmed. Two peaks representing the isomers (cis and trans) of 4-PrChOH were observed by GC-MS. | ||
 | ||
| Fig. 5 ECH of 4-PrPhOMe and 4-PrPhOH. Product yields, substrate conversion and faradaic efficiency. Experimental conditions: current = 60 mA; (mol e−)/(mol substrate) = 20 (4-PrPhOH and 4-PrPhOMe); temperature = 60 °C. Error bars represent standard errors. | ||
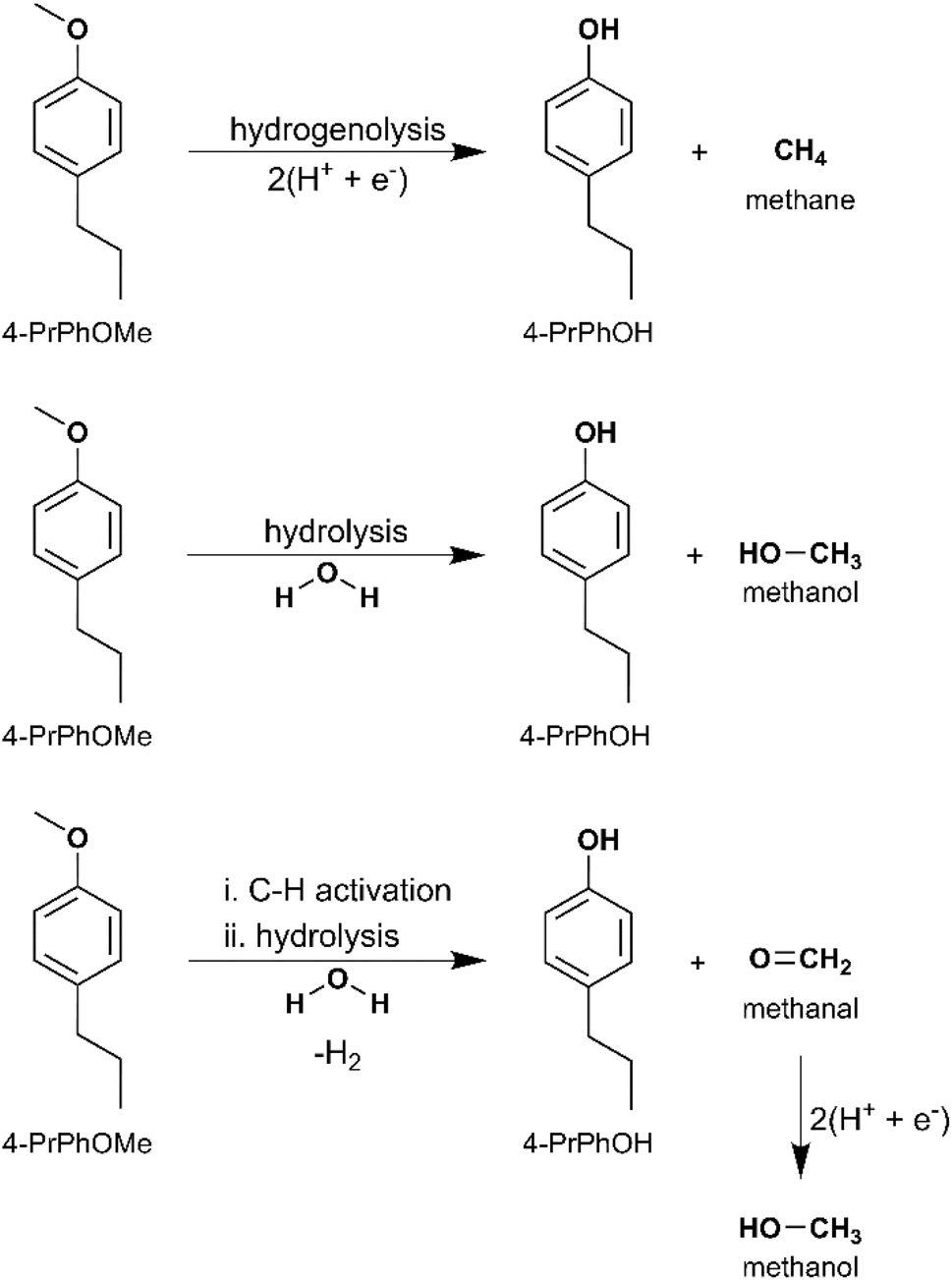 | ||
| Scheme 5 4-PrPhOMe ECH: demethylation pathways. | ||
The DFT calculations for guaiacol and anisole adsorption on Ru (0001) under vacuum indicate the following patterns. For guaiacol: C2, C4, C6 and Oa bind on top of 4 Ru atoms, Ob resides close to a hollow site and is tilted away from the Ru surface. For anisole: C2, C4, C6 and O bind on top of 4 Ru atoms, C7 resides close to a hollow site and is tilted away from the Ru surface.51 These adsorption patterns could explain the low selectivity of the demethylated product, notwithstanding the lower energy requirement for cleaving the methyl group. Note that while the reported BDEs based on homolytic cleavage provide insights into relative bond strength, catalytic cleavage events involve trade-offs with factors like binding energy on the metal surface and interactions with other species in solution. Additionally, homolytic BDEs do not necessarily track with more polar/ionic mechanisms.
The likely intermediates along the HYD pathway include 4-alkylcyclohexanol and 3-alkyl-1-methoxycyclohexane. Therefore, ECH of 4-MeChOH and 1-methoxy-4-methylcyclohexane (1-MeO-4-MeChH) was investigated in the present study. 1-MeO-4-MeChH was selected instead of 1-methoxy-3-methylcyclohexane since it could be readily procured. No MeChH formation was observed from both substrates, which confirms that the deoxygenation step precedes aromatic ring saturation during cycloalkane formation by 4-RGu ECH. The recoveries of the substrates are shown in Fig. 6. It has been proposed that phenol hydrogenation to ChOH proceeds via the formation of a cyclohexanone intermediate.52 Therefore, the ECH of 4-methylcyclohexanone (4-MeChO) was also investigated. The primary product observed after ECH was 4-MeChOH, along with trace amounts of MeChH (see Scheme 6). The formation of MeChH likely involves either a tautomerization step or a carbene intermediate, as no MeChH formation was detected during the ECH of 4-MeChOH.
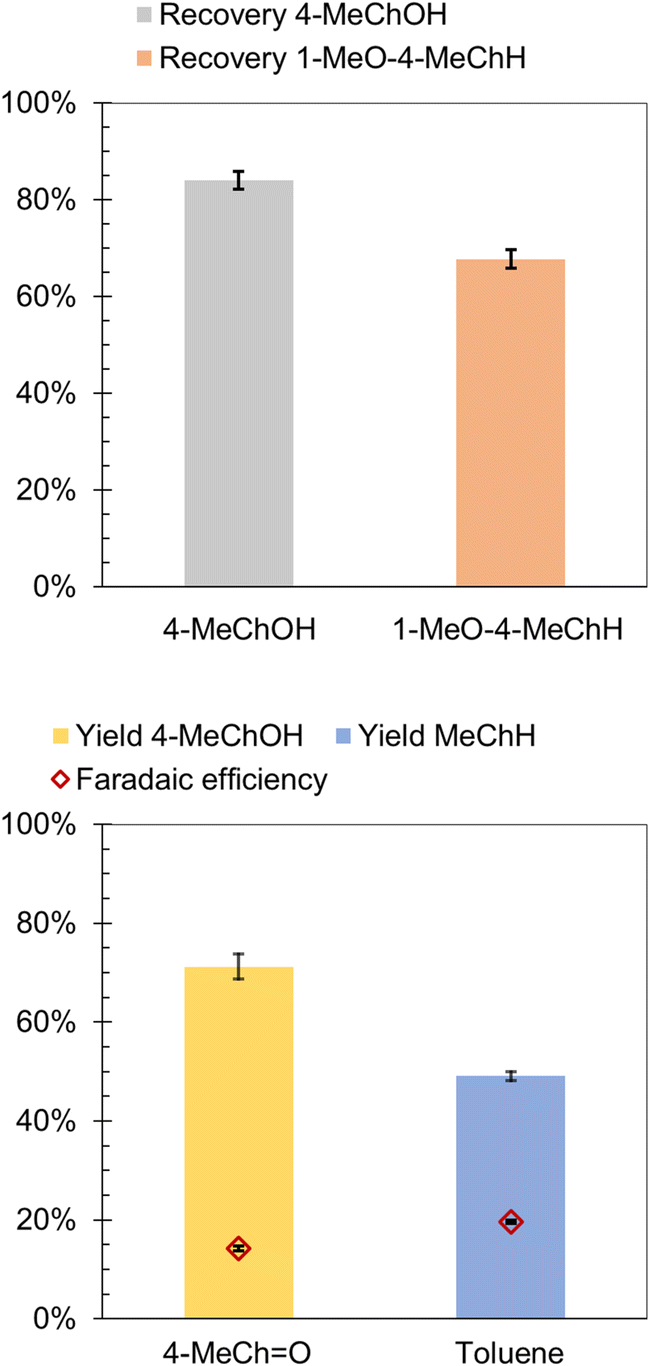 | ||
| Fig. 6 ECH of 1-MeO-4-MeChH, 4-MeChOH, 4-MeChO and toluene. (top) Substrate recovery. (bottom) Product yield and faradaic efficiency. Experimental conditions: current = 60 mA; (mol e−)/(mol substrate) = 10 (1-MeO-MeChH, 4-MeChOH and 4-MeChO) and 15 (toluene); temperature = 50 °C. Error bars represent standard errors. | ||
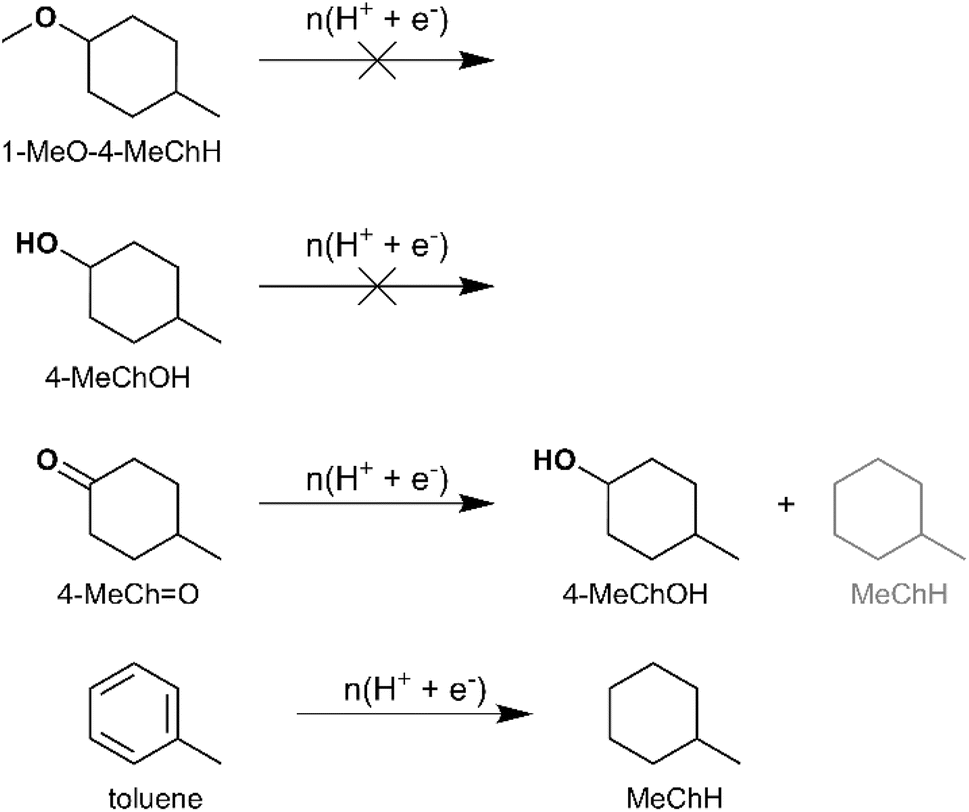 | ||
| Scheme 6 Products observed for ECH of 4-MeChO and toluene. No product formation by ECH was obtained from 1-MeO-4-MeChH and 4-MeChOH. Yield quantification was not performed for compounds in grey. Two peaks representing the isomers (cis and trans) of 4-MeChOH were observed by GC-MS. | ||
The final step in cycloalkane formation along a potential DDO or TAU pathway involves the saturation of an aromatic hydrocarbon. This reaction was confirmed to occur via an electrocatalytic pathway by performing toluene ECH, which yielded MeChH (see Scheme 6 and Fig. 6).
Overall, substrates with saturated rings did not undergo HDO during ECH, indicating that HDO occurred via the DDO or TAU pathway during 4-RGu ECH. Notably, operando Raman experiments by Page et al. have indicated that phenol HDO on RuPt-based electrocatalysts proceeds via a ketone intermediate,31 suggesting a preference for the TAU pathway. Moreover, for oxygenated aromatics, HDO and aromatic ring saturation are parallel reactions. Finally, it was demonstrated that the removal of the methoxy group occurred more readily than that of the hydroxy group, suggesting that demethoxylation preceded dehydroxylation during HDO by ECH. A reaction network highlighting the probable pathways leading to the formation of various products observed during 4-RGu ECH is shown in Scheme 7. Future work focusing on liquid hydrocarbon fuel production by ECH must examine suitable modifications to reaction conditions and electrocatalysts to improve selectivity towards HDO reactions. Alternatively, valorization pathways for 4-alkylcyclohexanols must be explored. Liu et al. have demonstrated that alkylation of oxygenated aromatics (e.g., phenol, guaiacol, etc.) with 4-alkylcyclohexanols produced dimers with C–C linkages.53 The higher molecular weight bicyclic alkanes produced by electrocatalytic or thermocatalytic HDO of these dimers could be suitable candidates for blending into jet fuels.
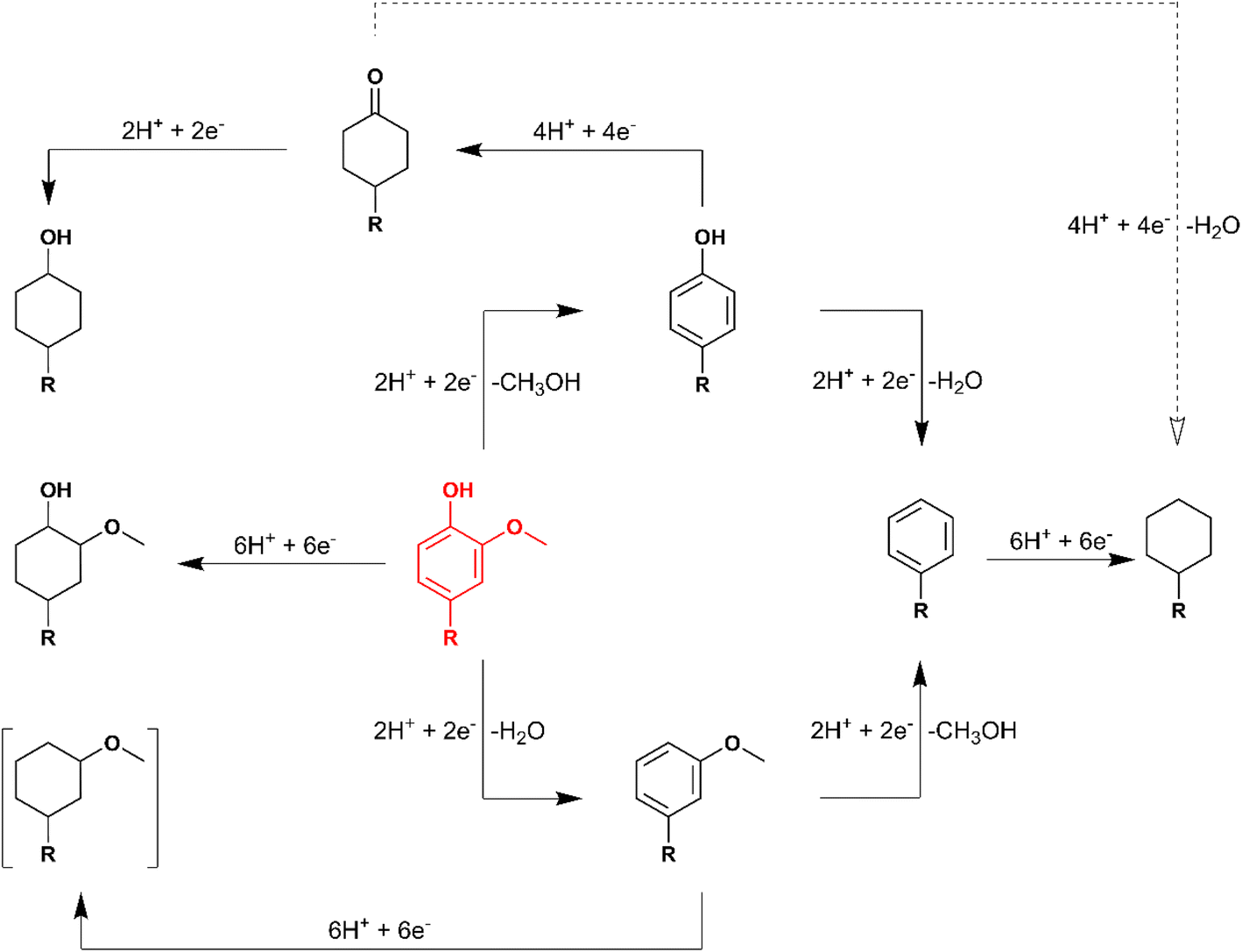 | ||
| Scheme 7 Proposed reaction network for ECH of 4-alkylguaiacol (in red). The dashed arrow denotes that the reaction yields only trace quantities of the product. | ||
Guaiacyl vs. syringyl moieties: implication for ECH reactions
Lignin is primarily composed of p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units. In grasses, lignin is made up of H, G, and S units, whereas softwood lignin is essentially composed of G units, and hardwood lignin is composed of G and S units.54 Consequently, fast pyrolysis and RCF of lignin from grasses and hardwoods result in oils containing compounds with syringyl units.55–58 Syringyl compounds have a higher oxygen to carbon ratio compared to analogous guaiacyl compounds due to the additional methoxy substituent on the aromatic ring. 4-Allylsyringol (4-AySy) [IUPAC: 2,6-dimethoxy-4-prop-2-enylphenol] was selected as a model syringyl compound in the present study. 4-AySy ECH on the Ru0.75Pt0.25/ACC electrocatalyst resulted in the formation of 4-propylsyringol (4-PrSy), 2-MeO-4-PrChOH, 4-PrChOH, and PrChH along with traces of 4-PrGu and 4-PrChO (see Scheme 8). The mass spectra of the unassigned peaks on the chromatogram indicated formation of 1-MeO-3-PrChH and 1,3-dimethoxy-5-propylbenzene; however, their identities could not be verified due to non-availability of reference mass spectra or neat standards; thus, the yields were not quantified. The complete conversion of 4-AySy and the absence of the allyl functionality in the products indicate that saturation of the olefinic group on the allyl chain occurs in the initial stages of the batch reaction process. The yields of PrChH and 4-PrChOH from 4-AySy ECH were significantly lower than those obtained from eugenol and 4-PrGu ECH (see Fig. 7), notwithstanding the passage of proportionately higher charge through the circuit ((mol e−)/(mol substrate) was 25 for 4-PrGu, 30 for eugenol, and 35 for 4-AySy). The low PrChH yield suggests that the additional methoxy group in 4-AySy decreases the rate of the HDO reactions. A similar effect has been observed in other studies that compared ECH of guaiacyl and syringyl compounds.17,18,49
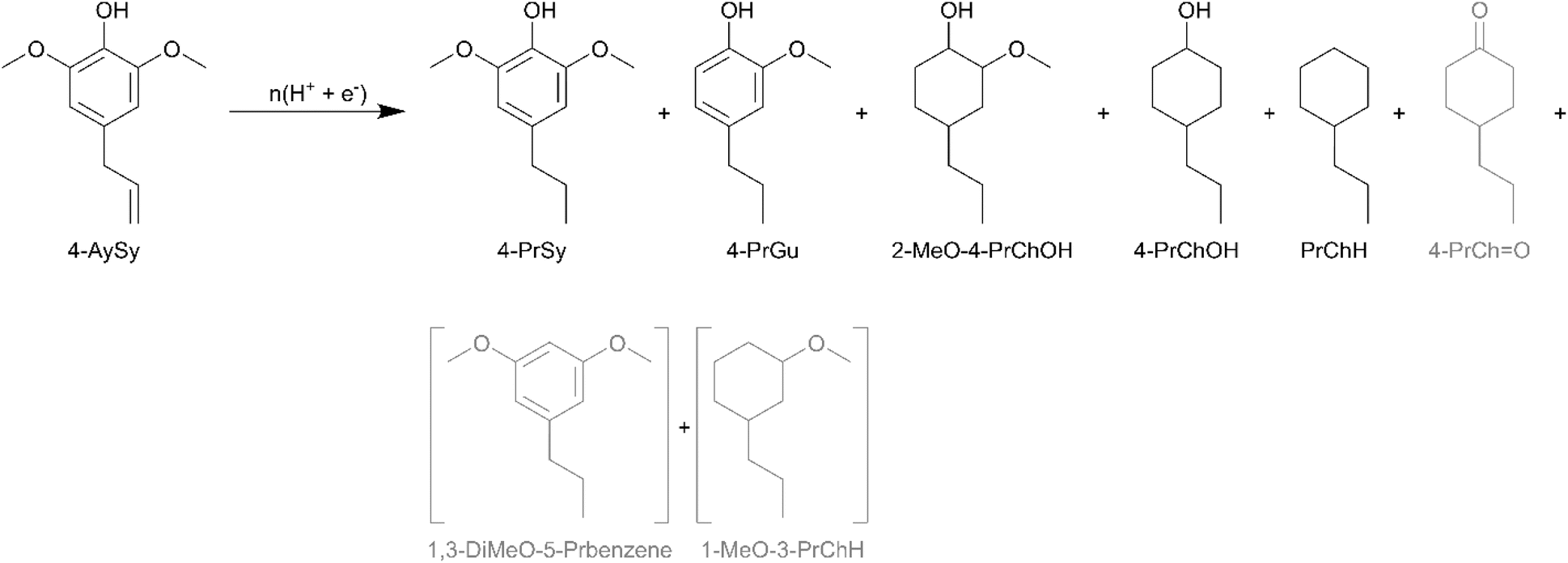 | ||
| Scheme 8 Products observed for ECH of 4-AySy. Yield quantification was not performed for compounds in grey. Yield quantification for 4-PrSy was performed by assuming it had the same response factor as 4-AySy. The formation of compounds enclosed in brackets is inferred but their identity was not confirmed. Two peaks representing the isomers (cis and trans) of 4-PrChOH were observed by GC-MS. Three peaks representing the isomers of 2-MeO-4-PrChOH were observed by GC-MS, but quantitation was performed only for the two peaks observed by GC-MS for the external standard. | ||
 | ||
| Fig. 7 ECH of 4-PrGu, eugenol and 4-AySy. Product yields, substrate conversion and faradaic efficiency. Experimental conditions: current = 60 mA; (mol e−)/(mol substrate) = 25 (4-PrGu), 30 (eugenol) and 35 (4-AySy); temperature = 60 °C. Error bars represent standard errors. | ||
ECH of di-p-tolyl ether
The 4-O-5 linkage is the strongest interunit ether linkage in lignin.59,60 Analytical pyrolysis of dimer models with a 4-O-5 linkage indicates resistance to thermal cracking. Garedew et al. demonstrated that 2-phenoxyphenol did not undergo pyrolytic cleavage at 650 °C.61 Meanwhile, Custodis et al. found that diphenyl ether pyrolysis at temperatures above 650 °C resulted in the formation of recombination products containing a 4-O-5 linkage with selectivity greater than 20% (up to ∼70%).62 Therefore, it is of interest to investigate the electrocatalytic reductive cleavage of the 4-O-5 linkage. In this regard, di-p-tolyl ether (Tol2O) [IUPAC: 1-Methyl-4-(4-methylphenoxy)benzene] was selected for investigation in the present study. The products observed upon ECH on Ru0.75Pt0.25/ACC include MeChH and 4-MeChOH, along with trace quantities of 4-MeCHO, indicating cleavage of the 4-O-5 linkage by an electrocatalytic pathway (see Scheme 9 and Table 3). The mass spectra of the unassigned peaks on the chromatogram indicated formation of dimer products with saturated rings (di-4-methylcyclohexyl ether and 4-methylphenyl-4-methylcyclohexyl ether); however, their identity could not be verified due to non-availability of reference mass spectra or neat standards. Therefore, the yields were not quantified. Garedew et al. demonstrated that the presence of a strong electron-donating substituent on the aromatic ring activated the ether oxygen for electrocatalytic C–O bond cleavage, based on a comparison of substrate conversion and cyclohexanol yield during the ECH of 3-phenoxyphenol and 3-phenoxytoluene.61 Importantly, the present study demonstrates the efficacy of the Ru0.75Pt0.25/ACC electrocatalyst in achieving C–O bond cleavage in the absence of a strong electron-donating group. Song et al. have demonstrated the formation of 4-methylphenol (4-MePhOH) and toluene, in addition to the products reported in the present study, from Tol2O ECH on Rh/C.63 It was proposed that C–O cleavage occurred via two distinct pathways: hydrolysis and hydrogenolysis. Song et al. inferred the occurrence of hydrolysis reactions based on the difference in yields of MeChH and 4-MeChOH, given that 4-MePhOH did not undergo HDO on Rh/C. The pathway for C–O cleavage on the Ru0.75Pt0.25/ACC electrocatalyst in the present study could not be similarly inferred. However, the higher yield of MeChH compared to 4-MeChOH suggests that the 4-MePhOH produced by C–O cleavage undergoes both HDO and aromatic ring saturation to form MeChH.
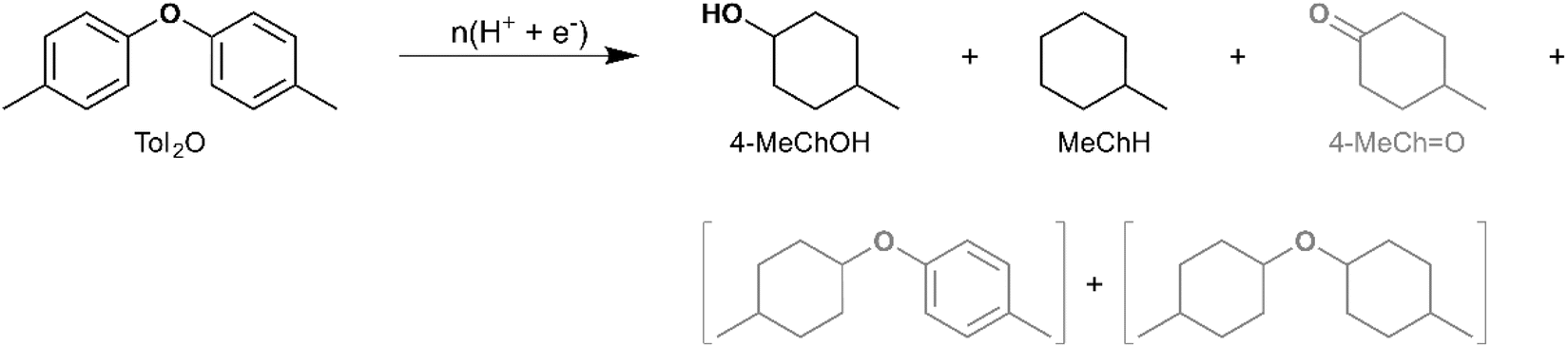 | ||
| Scheme 9 Products observed for ECH of Tol2O. Yield quantification was not performed for compounds in grey. The formation of compounds enclosed in brackets is inferred, but their identity was not confirmed. Two peaks representing the isomers (cis and trans) of 4-MeChOH were observed by GC-MS. | ||
| Conversion Tol2O | Yield 4-MeChOH | Yield MeChH | Mole balance | Faradaic efficiency |
|---|---|---|---|---|
| 78.0% (±0.6%) | 38.4% (0.0%) | 46.7% (±2.5%) | 64.6% (±0.6%) | 10.2% (±0.3%) |
Overall, the low faradaic efficiencies (<30%) observed in the present study indicate that a significant fraction of the current was diverted towards hydrogen gas formation. The evolution of small bubbles from the electrocatalyst surface, especially during the latter stages of the ECH trials, provided visual confirmation of hydrogen gas formation. While H2 is valuable and could be captured and utilized for thermocatalytic HDO of compounds such as alkylcyclohexanols, its formation during ECH may pose safety concerns. Optimizing the catholyte solution phenolic compound concentration and the ECH run time could improve the faradaic efficiency. Additionally, optimizing the operating potential of the cathode is crucial for scaling up the ECH of phenolic compounds. This optimization would depend, in part, on the onset potentials of the rate-determining step (hydrodeoxygenation or aromatic ring saturation) and the side reaction (hydrogen evolution), enabling the calculation of power requirements and overall process efficiency. The findings of the present study, combined with proposed future research, would inform technoeconomic feasibility assessments of electrocatalytic pathways for producing jet fuel blendstocks.
Conclusions
ECH of oxygenated aromatics was explored as a mild technique for converting substrates derived from lignocellulosic biomass or lignin to cycloalkanes. The bimetallic electrocatalysts (RuPt/ACC) exhibited superior performance compared to the monometallic electrocatalysts (Ru/ACC and Pt/ACC) for the ECH of 4-propylguaiacol. 4-Alkylguaiacol ECH yielded moderate quantities of alkylcyclohexane, with 4-alkylcyclohexanol as the primary side product. The ECH of eugenol and 4-allylsyringol resulted in comparatively lower cycloalkane yields, attributed to the inhibition of HDO reactions by the allyl substituent in eugenol and the additional methoxy substituent in 4-allylsyringol. A reaction network for ECH of 4-alkylguaiacols was developed by investigating the ECH of likely intermediates and side products. It was observed that demethoxylation occurred more readily than dehydroxylation. Moreover, for oxygenated aromatics, HDO and aromatic ring saturation are parallel reactions. Although the HDO mechanism prevalent during ECH was not determined, the hydrogenation-deoxygenation pathway, where complete aromatic ring saturation precedes the deoxygenation reactions, was rejected. Finally, the utility of ECH in cleaving ether linkages, like those in lignin, was demonstrated using a model compound, di-p-tolyl ether, which contains a 4-O-5 type linkage.Data availability
The data supporting this study have been included in the article and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Science Foundation under award number 2055068. Dr Saffron's contribution was supported in part by the USDA National Institute of Food and Agriculture, Hatch Project 1018335, and Michigan State University AgBioResearch. Elemental analysis (ICP-OES) was performed at the Michigan State University Quantitative Bio-Element Analysis and Mapping (QBEAM) Center. The authors warmly acknowledge the assistance provided by Abigail Vanderberg (SEM-EDS) and Dr Aaron Sue (ICP-OES).References
- EPA, Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990-2021, Report EPA 430-R-23-002, U.S. Environmental Protection Agency, 2023 Search PubMed.
- S. Dhakal, J. C. Minx, A. Abdel-Aziz, M. J. Figueroa Meza, K. Hubacek, I. G. C. Jonckheere, Y.-G. Kim, G. F. Nemet, S. Pachauri, X. C. Tan and T. Wiedmann, 2022, Emissions Trends and Drivers, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2023 Search PubMed.
- P.L. 110-140, Energy Independence and Security Act of 2007, United States, 2007 Search PubMed.
- P. Bajpai, in Pretreatment of Lignocellulosic Biomass for Biofuel Production, ed. P. Bajpai, Springer Singapore, Singapore, 2016, pp. 7–12, DOI:10.1007/978-981-10-0687-6_2.
- F. Stankovikj, A. G. Mcdonald, G. L. Helms and M. Garcia-Perez, Energy Fuels, 2016, 30, 6505–6524 CrossRef.
- E. Cooreman, T. Vangeel, K. Van Aelst, J. Van Aelst, J. Lauwaert, J. W. Thybaut, S. Van Den Bosch and B. F. Sels, Ind. Eng. Chem. Res., 2020, 59, 17035–17045 CrossRef CAS.
- S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- R. C. Striebich, L. M. Shafer, R. K. Adams, Z. J. West, M. J. DeWitt and S. Zabarnick, Energy Fuels, 2014, 28, 5696–5706 CrossRef CAS.
- J. T. Edwards, in 55th AIAA Aerospace Sciences Meeting, American Institute of Aeronautics and Astronautics, 2017, DOI:10.2514/6.2017-0146.
- J. A. Muldoon and B. G. Harvey, ChemSusChem, 2020, 13, 5777–5807 CrossRef CAS.
- J. Holladay, Z. Abdullah and J. Heyne, Sustainable Aviation Fuel: Review of Technical Pathways, 2020 Search PubMed.
- IEA, Global Hydrogen Review 2023, International Energy Agency, 2023 Search PubMed.
- B. Zhao, Q. X. Guo and Y. Fu, Electrochemistry, 2014, 82, 954–959 CrossRef CAS.
- W. Liu, W. Q. You, Y. T. Gong and Y. L. Deng, Energy Environ. Sci., 2020, 13, 917–927 RSC.
- Y. P. Wijaya, K. J. Smith, C. S. Kim and E. L. Gyenge, Green Chem., 2022, 24, 7469–7480 RSC.
- Q. L. Zhai, S. M. Han, C. Y. Hse, J. C. Jiang and J. M. Xu, Fuel Process. Technol., 2022, 227, 107109 CrossRef CAS.
- S. M. Han, X. L. Zhang, R. Z. Wang, K. Wang, J. C. Jiang and J. M. Xu, Chem. Eng. J., 2023, 452, 139299 CrossRef CAS.
- M. Garedew, D. Young-Farhat, J. E. Jackson and C. M. Saffron, ACS Sustainable Chem. Eng., 2019, 7, 8375–8386 CrossRef CAS.
- Y. Zhou, Y. Gao, X. Zhong, W. Jiang, Y. Liang, P. Niu, M. Li, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1807651 CrossRef.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, in Stud. Surf. Sci. Catal., ed. P. L. Llewellyn, F. Rodriquez-Reinoso, J. Rouqerol and N. Seaton, Elsevier, 2007, vol. 160, pp. 49–56 Search PubMed.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by Powders and Porous Solids, Academic Press, London, 1999 Search PubMed.
- S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, in Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Springer Netherlands, Dordrecht, 2004, pp. 129–156 Search PubMed.
- T. Suoranta, M. Niemelä and P. Perämäki, Talanta, 2014, 119, 425–429 CrossRef CAS PubMed.
- M. Balcerzak, Crit. Rev. Anal. Chem., 2002, 32, 181–226 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- J. Zhang, J. Sun and Y. Wang, Green Chem., 2020, 22, 1072–1098 RSC.
- A. Robinson, G. A. Ferguson, J. R. Gallagher, S. Cheah, G. T. Beckham, J. A. Schaidle, J. E. Hensley and J. W. Medlin, ACS Catal., 2016, 6, 4356–4368 CrossRef CAS.
- J. M. Sun, A. M. Karim, H. Zhang, L. Kovarik, X. H. S. Li, A. J. Hensley, J. S. McEwen and Y. Wang, J. Catal., 2013, 306, 47–57 CrossRef CAS.
- B. K. Jung, J. Lee, J. M. Ha, H. Lee, D. J. Suh, C. H. Jun and J. Jae, Catal. Today, 2018, 303, 191–199 CrossRef.
- M. Kim, J. M. Ha, K. Y. Lee and J. Jae, Catal. Commun., 2016, 86, 113–118 CrossRef CAS.
- J. R. Page, A. Pophali, T. Kim, J. A. Lopez-Ruiz, S. Bliznakov and J. A. Valla, Catal. Sci. Technol., 2024, 14, 5559–5573 RSC.
- A. Fukazawa, K. Takano, Y. Matsumura, K. Nagasawa, S. Mitsushima and M. Atobe, Bull. Chem. Soc. Jpn., 2018, 91, 897–899 CrossRef CAS.
- A. Fukazawa, Y. Shimizu, N. Shida and M. Atobe, Org. Biomol. Chem., 2021, 19, 7363–7368 RSC.
- Y. Du, X. Chen, W. L. Shen, H. B. Liu, M. Fang, J. X. Liu and C. H. Liang, Green Chem., 2023, 25, 5489–5500 RSC.
- M. Huš, A. Bjelić, M. Grilc and B. Likozar, J. Catal., 2018, 358, 8–18 CrossRef.
- L. Nie, P. M. de Souza, F. B. Noronha, W. An, T. Sooknoi and D. E. Resasco, J. Mol. Catal. A: Chem., 2014, 388, 47–55 CrossRef.
- Q. Tan, G. Wang, L. Nie, A. Dinse, C. Buda, J. Shabaker and D. E. Resasco, ACS Catal., 2015, 5, 6271–6283 CrossRef CAS.
- J. H. Zhang, J. M. Sun, B. Sudduth, X. P. Hernandez and Y. Wang, Catal. Today, 2020, 339, 305–311 CrossRef CAS.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen and A. D. Jensen, ACS Catal., 2013, 3, 1774–1785 CrossRef CAS.
- F. E. Massoth, P. Politzer, M. C. Concha, J. S. Murray, J. Jakowski and J. Simons, J. Phys. Chem. B, 2006, 110, 14283–14291 CrossRef CAS.
- J. Y. He, C. Zhao and J. A. Lercher, J. Catal., 2014, 309, 362–375 CrossRef CAS.
- X. Wang, M. Arai, Q. Wu, C. Zhang and F. Zhao, Green Chem., 2020, 22, 8140–8168 RSC.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 CrossRef CAS PubMed.
- C. Zhao and J. A. Lercher, ChemCatChem, 2012, 4, 64–68 CrossRef CAS.
- A. J. Foster, P. T. M. Do and R. F. Lobo, Top. Catal., 2012, 55, 118–128 CrossRef CAS.
- Y. P. Wijaya, T. Grossmann-Neuhaeusler, R. D. Dhewangga Putra, K. J. Smith, C. S. Kim and E. L. Gyenge, ChemSusChem, 2020, 13, 629–639 CrossRef CAS PubMed.
- Y. P. Wijaya, R. D. D. Putra, K. J. Smith, C. S. Kim and E. L. Gyenge, ACS Sustainable Chem. Eng., 2021, 9, 13164–13175 CrossRef CAS.
- Z. Li, M. Garedew, C. H. Lam, J. E. Jackson, D. J. Miller and C. M. Saffron, Green Chem., 2012, 14, 2540 RSC.
- C. H. Lam, C. B. Lowe, Z. Li, K. N. Longe, J. T. Rayburn, M. A. Caldwell, C. E. Houdek, J. B. Maguire, C. M. Saffron, D. J. Miller and J. E. Jackson, Green Chem., 2015, 17, 601–609 RSC.
- J. Shangguan, A. J. R. Hensley, M. V. Gradiski, N. Pfriem, J.-S. Mcewen, R. H. Morris and Y.-H. C. Chin, ACS Catal., 2020, 10, 12310–12332 CrossRef CAS.
- J. Lu and A. Heyden, J. Catal., 2015, 321, 39–50 CrossRef CAS.
- M. Gao, H. Tan, P. Zhu, J. Zhang, H. Wang, X. Liu and Z. Zheng, Appl. Surf. Sci., 2021, 558, 149880 CrossRef CAS.
- Y. Liu, G. Cheng, E. Baráth, H. Shi and J. A. Lercher, Appl. Catal., B, 2021, 281, 119424 CrossRef CAS.
- E. Dorrestijn, L. J. J. Laarhoven, I. W. C. E. Arends and P. Mulder, J. Anal. Appl. Pyrolysis, 2000, 54, 153–192 CrossRef CAS.
- S. Van Den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- J. M. Yuan, H. Li, L. P. Xiao, T. P. Wang, W. F. Ren, Q. Lu and R. C. Sun, Fuel, 2022, 319 Search PubMed.
- J. Hu, S. Wu, X. Jiang and R. Xiao, Energy Fuels, 2018, 32, 1843–1850 CrossRef CAS.
- O. Faix, D. Meier and I. Grobe, J. Anal. Appl. Pyrolysis, 1987, 11, 403–416 CrossRef CAS.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- R. W. Houston and N. H. Abdoulmoumine, RSC Adv., 2023, 13, 6181–6190 RSC.
- M. Garedew, D. Young-Farhat, S. Bhatia, P. Hao, J. E. Jackson and C. M. Saffron, Sustainable Energy Fuels, 2020, 4, 1340–1350 RSC.
- V. B. F. Custodis, P. Hemberger, Z. Ma and J. A. Van Bokhoven, J. Phys. Chem. B, 2014, 118, 8524–8531 CrossRef CAS PubMed.
- Y. Song, S. H. Chia, U. Sanyal, O. Y. Gutiérrez and J. A. Lercher, J. Catal., 2016, 344, 263–272 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01149j |
| This journal is © The Royal Society of Chemistry 2025 |